Strategies for Small Molecule Activation
| ✅ Paper Type: Free Essay | ✅ Subject: Chemistry |
| ✅ Wordcount: 3775 words | ✅ Published: 02 Aug 2017 |
Introduction
Developing novel strategies for small molecule activation is the core aim of catalysis reasearch. One approach that recently gained prominence in catalytic activation of organic molecules is photoredox catalysis. Visible light photoredox catalysis has risen to the interface of current organic chemistry as a remarkable way to facilitate single electron transfer (SET) processes with organic substrates upon photoexcitation1. This technique relies on the property of metal complexes and organic dyes to undergo SET with visible light2.
The commonly employed transition metal based photocatalyst used to harness the packets of energy carried by visible light are polypyridyl complexes of ruthenium and iridium, named as tris(2,2′-bipyridine)ruthenium (II) or Ru(bpy)32+ (Figure 1).
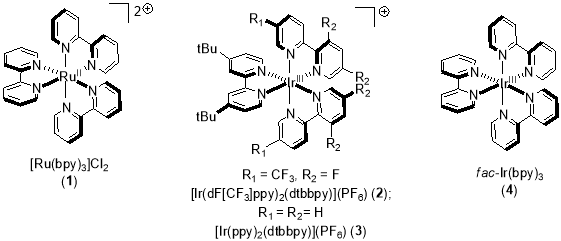
Figure 1. Structures of common transition metal photocatalysts.
These complexes give stable, long-lived excited states (for Ru (bpy)32+*, Ï„ = 110 ns)3 when irradiated with visible light of electromagnectic spectrum4. This relatively long lived excited state may allow bimolecular electron-transfer reactions through outer sphere transfer, both by the quenching of excited state photocatalyst and deactivation pathways5. The quenching can be accomplished in both oxidative and reductive ways (Figure 2), which offers this mode of catalysis flexibility.

Figure 2. Oxidative and reductive quenching cycles within photocatalysis.
Moreover, varying the metal (Ru, Ir, Cu, Cr,etc.) or ligands steer to foreseeable changes in redox potentials, enabling customization of the catalyst to one’s needs. In contrast to classical approaches these photochemical methods offer surprisingly mild conditions to radical reactions, as they typically operate at ambient temperature, utilize bench-stable reagents and display high degree of functional group tolerance.6
The extensive application of visible light photocatalysts have been recognized in the field of inorganic and materials chemistry. In particular, these catalysts have been found to be actively decomposing water into hydrogen and oxygen7 and reducing carbon dioxide to methane.8 Also, they have been employed in (i) as components in dye sensitized solar cells9 and organic light-emitting diodes,10 (ii) in polymerization reactions,11 and (iii) in photodynamic therapy.12
Until recently the reports of using these complexes as photocatalysts in organic synthesis were scarce. Their limited investigation was very surprising, as single electron, radical processes offer a unique pathway and reactivity to form C-C bond which are complementary to the closed shell, two electron processes.13 In the last decade detailed studies form the Yoon’s , MacMillan’ sand Stephenson’s groups have shown the application of Ru(bpy)32+ as photoredox catalyst to perform crucial C-C bond forming reactions such as [2+2] cycloaddition,14 α-alkylation of aldehydes15 and reductive dehalogenation of activated aryl halides.16 These quality work of above groups have rejuvenated the interests of many researchers in this field, triggering the diverse ideas into the utility of photoredox catalysis as conceptually novel approach to synthetic organic reaction development.
The application of visible light photoredox catalysis in organic synthesis revolves around its ability to engineer unusual bond constructions which are not easily formed by established protocols. For instance, overall neutral redox reactions can be performed by photoredox catalysis, as both the oxidant and reductants can be generated within the same reaction vessel. Visible light photoredox catalysis has been proved to be convenient in designing reactions, which needs gain and removal of electrons at disparate centres in a reaction mechanism. In contrast to these methods, others require stoichiometric quantity of both oxidant and reductants, which many times are incompatible with each other. Radical intermediates generated from single electron transfer (SET) events have been shown to have different reactivity patterns fundamentally different from those accessed through the ground state of catalyst.17 Harnessing these intermediates by means other than photoredox catalysis are often challenging or requires conditions which are incompatible with substrates.
It is noteworthy to mention, Ru (II) and Ir (III) based photocatalysts are widely used to generate radicals for use in a diverse range of radical reactions, and most of these reactions occur under mild conditions such as room temperature without the need of reactive radical generators (e.g., azobisisobutyronitrile (AIBN), BEt3), and toxic reagents (e.g., Bu3SnH), and in many cases, high temperature. The source of irradiation typically used are commercially available household light bulbs, which has significant advantage over specialized equipment employing high-energy ultraviolet (UV) light. Moreover, organic molecules generally do not show absorbance in visible region, so there is little probability of unwanted side reactions that might occur from the photoexcitation of the substrate itself. Even, the low photocatalyst loading of 1 mole % or less is sufficient enough to achieve high conversions. These all collectively have proven that visible light mediated photoredox catalysis to be a uniquely well-suited in designing safer and more sustainable strategies for synthesising more efficient materials and reducing waste streams. Further incentivizing the design and application of novel visible light-mediated methodologies toward both natural and non-natural scaffolds of interest to pharmaceutical and agrochemical domains.18
This review highlights the earlier work done on the use of Ru (II) and Ir (III) transition metal complexes as photoredox catalysts to promote C-C bond forming reactions in organic synthesis. Specifically, there is great emphasis on the applications of visible light photoredox catalysis which have enabled the total synthesis of natural products and related molecules, focusing on a range of powerful transformations that include: reductive coupling, indole functionalization, radical cascades, ATRA reactions, trifluoromethylation and selective C-O bond cleavage.
Reductive Dehalogenation
Reductive deahalogenation refers to process in which a C-X bond is reduced to a C-H bond where X denotes halogens. These classes of reactions have attracted attention of organic chemists all over the world due to its prime importance in rational organic synthesis. For instance a significant number of examples of these reactions can be found in nature, where enzymatic dehalogenation is performed by microorganism present in soil to check the concentration of lipophilic halogenated species.19 There has been a whole library of reducing systems developed to carry out reductive dehalogenation successfully, which practically guarantees the existence of specific reagents for specific substrate.
Organo-tin hydride has been the most used reagent in the past to perform reductive dehalogenation in laboratory as well as in field of synthesis, as it has been proven capable for both radical generation and kinetic radical trapping.20 By far, the system of tin hydride is tributyltin hydride (TBTH) – (AIBN) is the most utilized for radical-promoted dehalogenations of organic halides.21 However, there are three main problems in use of TBTH. First, toxicity of tin rule out its use in pharmaceutical synthesis. Second, there are lots of problem associated with the purification of reaction mixture from tributyltin residues. Third, TBTH is not a stable compound, even after careful storage it is likely to steadily decompose.22 It is the toxicity, that has almost precluded its use in a wide range of useful radical reactions in organic synthesis.
In recent years, the search for superior alternatives to TBTH has been the central goal of radical chemists. A replacement reagent needs to overcome all three problems mentioned above while at the same time an exhibiting similar reactivity and an ease of use. Earlier work of Fukuzumi and Tanaka focused on use of Ru(bpy)32+ as a photo redox catalyst to promote the reductive dehalogenation of phenacyl bromides23 and reductive dimerization of benzyl bromide24 respectively (Scheme 1), has shown that the application of visible light photoredox catalysis to access radicals can offer a promising solution to this problem.
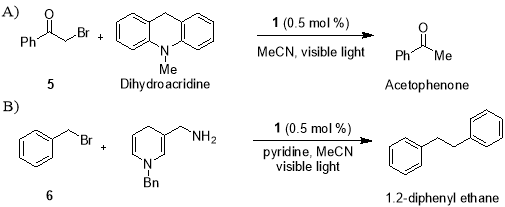
Scheme 1. Reductive dehalogenation of phenacyl bromide (A) and reductive dimerization of benzyl bromide (B).
But, it was the efforts of Narayanam and co-workers, focussed on developing the novel means for accessing radical chemistry while avoiding the toxicity and problems associated with tin hydride, has laid a milestone in development of a tin-free reductive dehalogenation systems (Scheme 2.).25
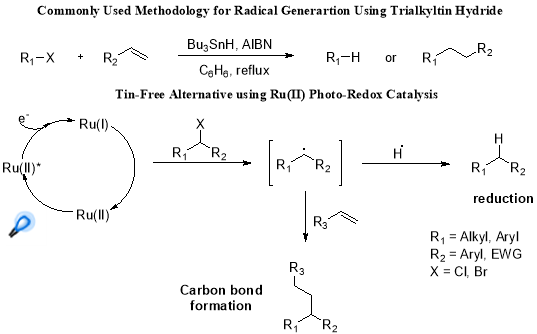
Scheme 2. Photoredox catalytic reduction and potential C-C bond formation.
In their primary investigation, Narayanam et al. used a system consisting Ru(bpy)32+ as a photocatalyst, iPr2NEt as major hydrogen atom source and visible light to successfully perform the reductive debromination. In the net transformation, the 3-bromopyrroloindoline (7) was reduced to pyrroloindoline (8) as single product, with the addition of Hantzsch ester or formic acid to the catalytic system produced debrominated product in >90% yield (Scheme 3).

Scheme 3. Initial attempt for reductive dehalogenation.
In further development of general tin-free visible light mediated dehalogenation protocol, a range of different activated alkyl bromides and chlorides were tested which afforded the corresponding dehalogenated product in good to excellent yield. Although, the un-activated aryl and alkenyl iodides were completely unreactive, as it was expected due to their exceptional negative reduction potentials  (-2.24 V Vs SCE for iodobenzene).26 The solution to this problem lied in the use of Ir(III) based photo-catalysts instead of Ru(II), which offered more reducing power than Ru(bpy)32+,and the dehalogenation of less activated alky, vinyl and aryl iodides with good functional group tolerance was achieved using oxidative quencing cycle of fac-Ir(bpy)3 (Scheme 4).27Â
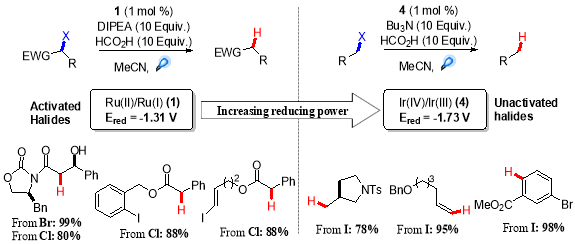
Scheme 4. Reductive dehalogenation of activated and unactivated halides.
Furst et al. used this practical strategy for reductive dehalogenation for a further development of more challenging intermolecular C-C bond forming protocols, which demonstrated an efficient way to promote intermolecular additions using visible light mediated photoredox catalysis. Furst et al. reported a facile coupling of indole with malonate radicals, as malonate-like motifs are common C2-subsitutents in bioactive indole based alkaloids such as actinophyllic acid (9) and undulifoline (10).
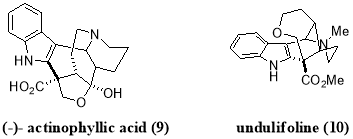
Using this procedure, an extensive range of indole and pyrrole derivatives were synthesized in good yields by employing (1) as the photocatalyst and N,N-diphenyl-4-methoxyaniline as the reductive quencher (Scheme 5).28 Further, this protocol was extended by Stephenson et al. to synthesize quaternary carbon centres adjacent to C2 of indole based alkaloids by employing more challenging tertiary malonate radicals.29 This transformation was accomplished by directly reducing bromomalonate (11) via oxidative quenching of more reducing fac-Ir(bpy)3 photocatalyst, providing targeted quaternary carbon centres in good to high yields (Scheme 6).
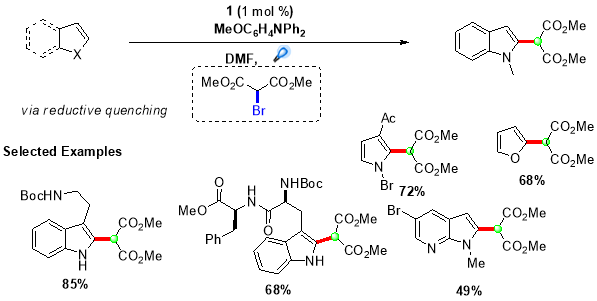
Scheme 5. Intermolecular radical addition of secondary radicals to electron-rich heterocycles
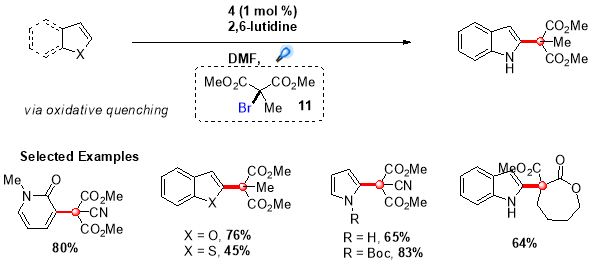
Scheme 6. Intermolecular radical addition of tertiary radicals to electron-rich heterocycles.
Atom Transfer Radical Addition (ATRA)
These transformations was first observed by Kharasch30 in 1940s, over the time atom transfer radical addition sparked the interest of organic chemists, as it offers the potential for uniquely efficient and economical approach for dual functionalization of olefins. This functionalization leaved a tremendous impact in organic chemistry, and have also found wide applications in industry and academic research. Similar to the intermolecular malonate-indole coupling mentioned above, these transformations are redox neutral, theoretically eliminating the need for additives, which in terms, reduces the likelihood of deleterious off-target reactivity.
The most important application of atom-transfer radical addition reactions is inclusion of fluorinated functional groups into molecules, as the addition of these groups has a strong impact on biological properties and bioavailability of bioactive compounds.31 In 2011 Stephenson, et al. for the first time reported visible light mediated ATRA reactions, proving this methodology as an efficient way to improve the overall performance of this kind of reaction compared to classic radical initiation conditions. This synthetic approach was effective for the preparation of a wide range of perflourohalogenated substrates from unactivated alkenes by using Ru(bpy)32+ as the photocatalyst combined with sodium ascorbate as an electron donor (Scheme 7).32
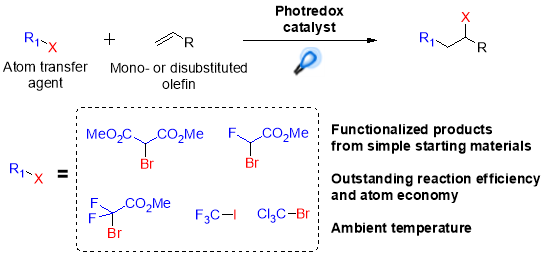
Scheme 7. Atom transfer radical addition mediated by photoredox catalyst.
A similar kind of transformation also providing halotrifluoromethylated product was reported by Han et al. (Scheme. 8)33 using triflouromethanesulfonyl chloride as the triflouromethyl source and visible light in presence of Ru (II) photocatalyst (1). Using this protocol, the variety of substrates including mono, di-, and tri-substituted unactivated alkenes went under trifluoromethylation in excellent yields.
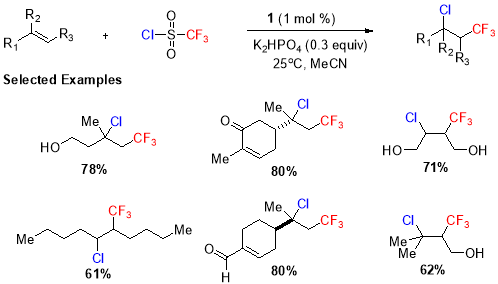
Scheme 8. Trifluoromethyl chlorination of disubstituted and internal alkenes.
Radical Cascades
Radical cascades are one of the most powerful tools for accessing complex structures in single step if substrate is stable under the for radical initiation conditions.34 One of the earliest examples of radical cascade was reported by Stokes et al.35 is intermolecular addition of Sn-radical to alkynes, he also studied the regioselectivity of vinyl radical cyclizations onto C=C double bond (Scheme 9). Cyclization cascades initiated by intermolecular addition of Sn radical to alkyne can be distinguished between reactions where tin-moiety retained in the final product with those where Sn radical essentially acted as a catalyst, which was later removed by the homolytic cleavage of labile C-Sn bond.
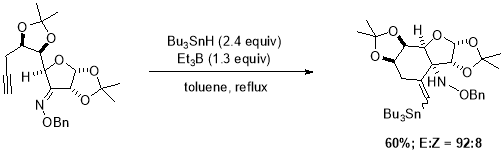
Scheme 9. Radical cyclization sequence, triggered by regioselective addition of tin radical.
Nowadays, because of the recognized toxicity associated with organotin compounds, the focus has been shifted toward the development of alternative tin-free and less environmentally problematic methods for radical cyclizations. Visible light photocatalysis has offered a powerful and sustainable tool for the development of new catalytic radical cascade reactions due their unique ability to facilitate formation of various reactive radicals and radical ions in mild and environmental friendly conditions. Various structurally diverse carbocycles and heterocycles from basic and readily available materials have been synthesis by using this protocol.
The augmentation of radical cascade cyclization and visible light photoredox catalysis approach has inspired radical chemists around the world to develop novel and efficient methods for synthesis of important heterocyclic motif that are prevalent in nature products exhibiting a wide range of bioactivites. One highly effective method for radical cascade, generating tetracyclic fused ring was reported by Furst et al. where they used visible light mediated protocol to synthesize tetracycle from bromomalonate and tricyclic compound from alkyne in good yields as a single diastereomers36 (Scheme 10.).
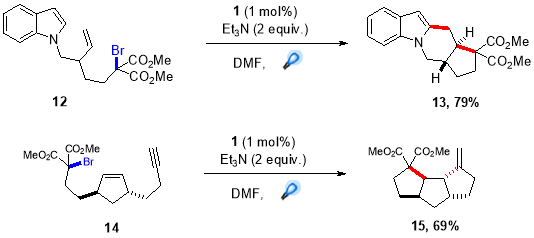
Scheme 10. Intramolecular radical cascades.
Xiao et al. further extended the application of visible light mediated radical cascade reactions in synthesis chromam-2ones and dihydroquinoline-2-ones based scaffolds, as these are omnipresent components in biologically active natural products and pharmaceutical drugs37. They reported a new type of radical cascade reaction between photogenerated α-amino radicals and acyloyl ester- and acrylamide-tethered aroylhyrazones.38 (Scheme 11).

Scheme 11. Photoredox catalyzed radical cascade reaction of α-amino radicals.
In addition, they developed an oxidant free N-radical cascade reaction of β, γ-unsaturated hydrazones by incorporating visible light photoredox and cobalt catalysis to obtain dihydropyrazole-fused benzosultams (Scheme 12),39 that has never been reported previuosly.

Scheme 12. Visible light photocatalytic N-radical cascade reaction of benzosultam synthesis.
Recently, Xu et al. devised a valuable cascade annulation by generating acyl radicals from abundant acyl chlorides under visible light mediated photoredox catalysis which then started a cascade cyclization of 1,7 enynes (Scheme 13).40

Scheme 13. Visible light induced cascade cyclization of 1,7-eynes with acyl chlorides.
Applications in Total Synthesis
In the history of organic synthesis, indole based alkaloids grabbed much more attention because of their abundance in natural products and biologically active compounds, and they have always been interesting and challenging synthetic targets. The unique ability of visible light mediated photoredox catalysis in forming key C-C bond granted access to numerous applied intermediates that facilitated synthesis of these diverse natural products.
In 2011, Stephenson and co-workers reported the asymmetric synthesis of (+)-gliocladin C (21), a natural product with interesting cytotoxic activity (Scheme 14.)41 starting from L-tryptophan, the important intermediate C3 bromopyrroloindoline (17) was synthetically prepared by standard transformations using Boc-D-tyrptophan methyl ester (16). The vital step in the synthesis was the formation of C-C coupled intermediate (18), which was accomplished by reductive dehalogenation-arylation process triggered by blue light irradiation on substrate in the presence of aldehyde (22), photocatalyst (1) and NBu3 as a quencher. This intermediate was converted into natural product in 7 high-yielding steps, which was more efficient than the previous reported 21-step structural synthesis of (21) starting isatin with and overall yield of ~ 4%.42
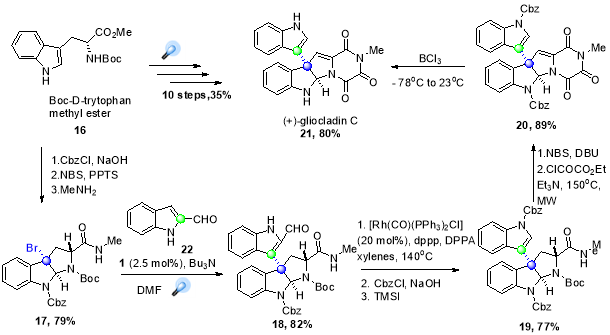
Scheme 14. Total synthesis of (+)-gliocladin C.
Another more recent example is the synthesis of biologically active alkaloids drimentines A, F and G (Scheme 15.).43 by Li and co-workers utilising reductive C-C bond forming strategy. In this example, the heterocycle (25) was coupled with acceptor (24) by intermolecular radical 1,4-addition to generate the important intermediate (26), which facilitated the product (27) -(29) in good yields.

Scheme 15. Total synthesis of drimentines A, F, G
Targeting Pharmaceutically Relevant Scaffolds
The unique capabilities of photoredox catalysis is an access to variety of fluoroalkyl radical species at late stage modification of therapeutic leads. Fluorinated functional groups (trifluoromethyl group in particular) have become increasingly popular over the decades44, because these motifs have dramatic on the molecules’ physiochemical properties, making them more selective, increasing their efficacy, or making them easier to adminster. Photoredox catalysis can provide an approach tailored on industrial scales by using abundantly available CF3 sources and eliminating the need of pre-functionalized substrates. This chemistry was readily translated to multigram scales for a number of substrates, one most important example of this strategy is the synthesis of trifluoromethylated 2-chloropyridine (32) (Scheme. 16) a vital synthon in production of anti-infective agents at Boehringer Ingelheim Pharmaceuticals Inc.45
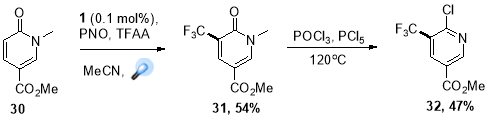
Scheme 16. Preparation of Boehringer-Ingelheim intermediate.
A novel redox system comprising pyridine N-oxide and trifluoroacetic acid was designed by Beatty at el. where C-C activation was achieved by pyridine N-oxide, a “redox trigger”, which could in situ generate modified trifluoroacetate shifting the redox potential of trifluoroacetate lower, within the reach of Ru(bpy)32+ photoredox catalyst.46
ORL-1 Antagonist Intermediate
Opioid receptor-like 1 (ORL-1) antagonist which is currently under the development for the cure of depression and obesity,47 has a gem-difluorobenzyl functional group around the spirocyclic piperidine (34), the earlier synthetic route consisted a total of 8 steps starting from (33) with an overall yield of 28%.48 Futhermore, this sequence included AIBN initiated radical bromination, and most challenging step was the benzylic fluorination by using 2.6 equiv. of Deoxo-Fluor 9 (specialised fluorinated reagent) as a fluoride source, which remained problematic as it required the use of pre-functionalized substrates through classical nucleophilic or electrophilic fluorination. Radical rearrangements reactions have demonstrated the strategic benefits in its synthesis when compared to this method.49
Visible light mediated radical Smiles rearrangement was developed to address the challenging synthesis of gem difluoro group ORL-1 antagonist from fluorinated thiophene (35), which could be produced from difluoro-ethanol from readily available corresponding ethyl ester (Scheme 17) reported by Douglas et al.50 This strategy has solved the problem of high number of steps and overcome the overall low yield and use of specialized fluorinated reagents. This new 5-step synthetic route eliminated the undesirable feature of previous synthetic route, the challenging benzylic defluorination could be accomplished by switching a key transformation to a C-C bond instead of a C-F bond formation.

Scheme 17. Previously reported route towards ORL-1 antagonists and new photochemical radical smiles rearrangement route.
Biofeedstock Processing
Biomass set itself aside from other renewable resources, since the energy it contains is stored in the form of chemical bonds, which allow biomass to be used for several purposes other than generating electricity and heat, such as liquid fuel and value-added chemicals. In particular, depolymerisation of lignin50, one of the most abundant feedstock for aromatic commodity compounds, which has attracted a lot of attention in recent years.
Lignin is a stable, branched biopolymer which is a part of the plant cell wall and is primarily responsible for providing rigidity and protection against environmental conditions. Primarily, it is composed of three different types of cinnamyl alcohols coupled together to produce a various array of motifs inside of the polymer chain (Scheme 18). The multiple connectivity and stability has hindered attempts to efficiently isolate value compounds through the degradative processing.51
The most sensible point of start in lignin degradation is β-O-4 linkage, as this is the most abundant (45-65%). Photoredox catalysis provides mild means of cleaving these critical bonds by a two-step procedure, which includes the selective oxidation of the alpha carbon followed by photochemical reductive cleavage.52

Scheme 18. Two steps protocol for degradation of lignin model system.
This strategy could be used for efficient degradation of a range of lignin model systems, isolation of the fragmentation products in excellent yields by employing photocatalyst 3 under the reductive quenching conditions.
Conclusions.
Photoredox catalysis with Ru (II) and Ir (I) metal complexes has recently received widespread attention as a tool for synthetic chemists, and it has been applied to the development of wide range of new C-C bond forming reactions. The utility of photoredox catalysis arises not form its ability to promote C-C bond formation, but rather from its ability to generate a diverse array of reactive via single-electron transfer. As shown, these species include electrophilic α-carbonyl radicals, tert-malonate radicals, α-amino radicals, acyl radicals and trifluoromethyl radicals. These intermediates have been used to develop reactions as varied as reductive dehalogenation, indole functionalization, atom transfer radical additions, radical cascades and Smiles rearrangement.
Also, photoredox catalysis has been proved as valuable tool for the synthesis of various biologically active compounds and their derivatives, as demonstrated by its application in the total synthesis of gliocladin C, drimenties A, F, G, and pharma relevant scaffolds. In each of these syntheses, simple and typically inert functionalities in the starting materials are transformed into reactive intermediates upon single electron transfer. These powerful transformations are not only redefining the synthetic strategies, but it has also changed the face of radical chemistry; a fundamental area in organic chemistry which mostly accessible using hazardous radical reagents. These robust class of reactions have inspired many researchers in designing and developing novel approaches to synthetic targets. The growth of visible light phototredox catalysis is not only significant on its own right, also bodes well for the future of organic synthesis.
References:
- Nicholls, T. P.; Leonori, D.; Bissember, A. C., Applications of visible light photoredox catalysis to the synthesis of natural products and related compounds. Natural Product Reports 2016, 33 (11), 1248-1254.
- James J. Douglas, J. D. N.; Kevin P. C., Enabling Novel Photoredox Reactivity via Photocatalyst Selection. Aldrichimica 2014, 47, 15-25.
- Kalyanasundaram, K., Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues. Coordination Chemistry Reviews 1982, 46, 159-244.
- Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A., Ru(II) polypyridine complexes: photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coordination Chemistry Reviews 1988, 84, 85-277.
- Juris, A.; Balzani, V.; Belser, P.; von Zelewsky, A., Characterization of the Excited State Properties of Some New Photosensitizers of the Ruthenium (Polypyridine) Family. Helvetica Chimica Acta 1981, 64 (7), 2175-2182.
- Tucker, J. W.; Stephenson, C. R. J., Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. The Journal of Organic Chemistry 2012, 77 (4), 1617-1622.
- (a) Graetzel, M., Artificial photosynthesis: water cleavage into hydrogen and oxygen by visible light. Accounts of Chemical Research 1981, 14 (12), 376-384.
(b) Meyer, T. J., Chemical approaches to artificial photosynthesis. Accounts of Chemical Research 1989, 22 (5), 163-170.
- Takeda, H.; Ishitani, O., Development of efficient photocatalytic systems for CO2 reduction using mononuclear and multinuclear metal complexes based on mechanistic studies. Coordination Chemistry Reviews 2010, 254 (3-4), 346-354.
- Kalyanasundaram, K.; Grätzel, M., Applications of functionalized transition metal complexes in photonic and optoelectronic devices. Coordination Chemistry Reviews 1998, 177 (1), 347-414.
- Lowry, M. S.; Bernhard, S., Synthetically Tailored Excited States: Phosphorescent, Cyclometalated Iridium(III) Complexes and Their Applications. Chemistry – A European Journal 2006, 12 (31), 7970-7977.
- (a) Lalevée, J.; Blanchard, N.; Tehfe, M.-A.; Morlet-Savary, F.; Fouassier, J. P., Green Bulb Light Source Induced Epoxy Cationic Polymerization under Air Using Tris(2,2′-bipyridine)ruthenium(II) and Silyl Radicals. Macromolecules 2010, 43 (24), 10191-10195.
(b) Fors, B. P.; Hawker, C. J., Control of a Living Radical Polymerization of Methacrylates by Light. Angewandte Chemie International Edition 2012, 51 (35), 8850-8853.
- Howerton, B. S.; Heidary, D. K.; Glazer, E. C., Strained Ruthenium Complexes Are Potent Light-Activated Anticancer Agents. Journal of the American Chem
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal