Freezing Point Depression Osmometer
| ✅ Paper Type: Free Essay | ✅ Subject: Chemistry |
| ✅ Wordcount: 3434 words | ✅ Published: 17 Aug 2017 |
1. Osmolality is a commonly used unit of measurement that represents the concentration of a solution as the total number of solutes per kilogram of pure solvent (mOsm/kg):
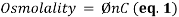
where Ø is the osmotic coefficient accounting for the degree of molecular dissociation; n is the number of particles left after the molecule dissociates in solvent, where n = 1 for non-electrolytes; and C is the molal concentration of the solution (moles/kg of water). As a variant of molality, only osmotically active particles that affect a solution’s osmotic pressure are considered. It is the number, rather than the size of type, of these solutes that controls the osmotic pull of a solution. Specifically, the presence of solute particles dilutes the solvent and restricts it to remain as a liquid as it is entropically favorable. Fittingly, the freezing point depresses proportionally with the increase in solute concentration since the temperature continues to drop instead of reaching a plateau during the process of crystallization as more pure solvent becomes separated from solution – leaving behind a smaller mass of liquid with higher solute concentration. Accordingly, the concentration of a solution can be determined by its relationship with this colligative property; thus, osmolality is really a measure of the chemical ac of water in an aqueous solution of dissolved particles. Overall, this term-independent of temperature and pressure-is used in medical laboratories (as opposed to osmolarity (mOsm/L) for bedside calculations) to describe the osmotic strength of bodily fluids as it can be easily attained by freezing point osmometry.
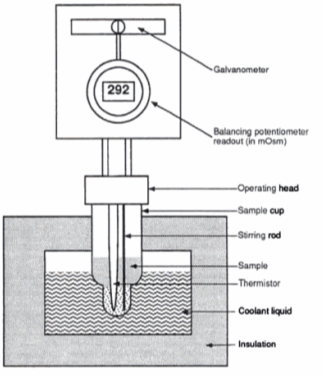
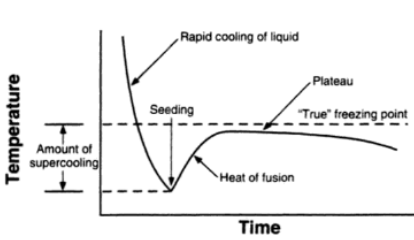
A freezing point depression osmometer quantifies the amount of osmotically important body fluid chemicals dissolved in blood serum by the relationship that 1 mole of particles decreases the freezing point of 1 kg of water by 1.86ï‚°C. This device is calibrated using standards within the osmolal range of interest (250-350 mOsm/kg for blood serum). Applied on a paper slide, the sample is inserted into an insulated-cooling module of circulating ethylene glycol and water refrigerants that chill the solution below its freezing point. An operating head then slides down on the sample container, immersing a thermistor temperature probe and stirring wire. Once the solvent molecules have aggregated and been supercooled, the stirring is set to vibrate more rapidly and aggressively to seed the solution with crystals, partially freezing it into a slush. During this liquid-to-solid phase transition, thermal energy is released into the solution as heat of fusion and proceeds until a temperature plateau that is slightly below the true freezing point is reached. The thermistor responds to this temperature change by altering its electrical resistance, thereby creating small variations in current – sensed by a galvanometer, which also detects the direction of current flow in the Wheatstone bridge that subsequently measures the unknown resistance. Lastly, a balancing potentiometer adjusts this resistance until the galvanometer returns to its null position of zero current, sequentially displaying the osmolality that is calculated by the following formula:
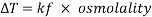
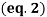
where kf is the cryoscopic constant (1.86 K⋅kg/mol) and ΔT is the temperature change. Diabetes mellitus is a chronic endocrine disease characterized by abnormally high serum glucose (>360 mOsm/kg). In a study conducted by Siervo et al., they measured blood serum osmolality levels in diabetic and non-diabetic older adults as an indicator of their hydration status, which is correlated to this disease. By using the Bland-Altman method to compare to a measured reference standard (via an osmometer), a successful serum osmolarity prediction formula-based on freezing point depression-was used to assess osmotically important chemicals in the control and study group:
Calculated Osmolarity = 1.86 Ã- (Na+ + K+) + 1.15 Ã- glucose + urea +14 (mmol/L) (eq.3)
With 79% sensitivity and 89% specificity, this equation serves as a first-stage screening method for diabetes diagnosis. Individuals with diabetes mellitus was shown to have higher serum osmolality levels (> 300 mOsm/kg) and glucose levels – characteristic of their dehydration state.
2. Fluorescence anisotropy describes the phenomenon that occurs when a fluorophore that has been excited with linearly polarized light emits fluorescence with unequal intensities along different polarization axes – as its absorption and emission transition moments lie along specific directions within its structure. The degree of this linear polarisation in its emission-resulting from photoselection of an optically isotropic sample-is described by its steady-state anisotropy:
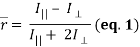
where I|| and I⊥ are relative intensities detected for emission that is parallel and perpendicular to the electric vector of linearly polarized incident light, respectively (where a non-zero 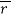 reveals a polarized emission). The denominator represents total fluorescence intensity (I) as it incorporates the three mutually orthogonal emission components, including the second perpendicular emission plane that sets anisotropy apart from polarization. By timeâ€resolved measurements, this quantifies depolarisation of fluorescence emission mainly caused by an energy transfer to another molecule of a different orientation or rotation (due to Brownian motion). Subsequently, enzyme-substrate binding constants and reaction kinetics can be studied since the rotational correlation time of a molecule would change, which is related to anisotropy by a Perrin equation – thus allowing for determination of its molecular size and mobility:
reveals a polarized emission). The denominator represents total fluorescence intensity (I) as it incorporates the three mutually orthogonal emission components, including the second perpendicular emission plane that sets anisotropy apart from polarization. By timeâ€resolved measurements, this quantifies depolarisation of fluorescence emission mainly caused by an energy transfer to another molecule of a different orientation or rotation (due to Brownian motion). Subsequently, enzyme-substrate binding constants and reaction kinetics can be studied since the rotational correlation time of a molecule would change, which is related to anisotropy by a Perrin equation – thus allowing for determination of its molecular size and mobility:
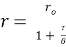 (eq. 2)
(eq. 2)
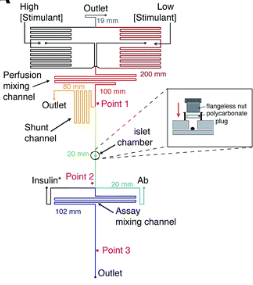
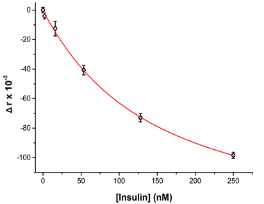
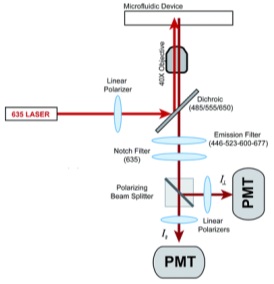
where ro is fundamental anisotropy of the fluorophore; Ï„ is the fluorescence lifetime; and θ is the rotational correlation time. The purpose of the study conducted by Schrell et al. was to develop a microfluidic biochemical test that can monitor insulin secretion dynamics upon glucose stimulation of single islets via interactions between insulin and its antibody. They were inspired by the fact that assays available today involves difficult separation systems that pose a challenge to non-specialized laboratories. This device can examine mechanisms behind abnormal secretions of metabolic diseases such as diabetes mellitus in real-time. Islets of Langerhans were isolated from male mice and incubated in RPMI-1640 media. A singlet islet was housed in a microfluidic chamber and stimulated with various levels of glucose from a gravity-based perfusion system. These channels have inputs for low and high glucose concentrations. Flowing from a top-to-bottom direction, most of the mixed perfusion solution is sent to waste through a shunt channel while a portion of it is directed to a sealed islet chamber. The total perfusate containing the islet secretions then combines with solutions of high affinity insulin antibodies (Ab) and Cy5-labeled insulin* as it passes the assay mixing channel – where both insulin and insulin* competitively bind to Ab. Since insulin* has a smaller rotational time compared to the Ab-insulin* complex, its emission is more depolarized and by measuring the bound/free ratio of the Ab-insulin* complex and free insulin*-via capillary or microfluidic electrophoresis-insight on insulin secretion dynamics can be gained as it is indirectly proportional to the amount of insulin in the sample. With a laser-induced fluorescence detection system, linearly polarized light from a 635-nm laser passes through a linear polarizer and reaches a dichroic mirror that focuses the beam towards the microfluidic channel (to excite the immunoassay mixture) via a microscope – which also collects the fluorescence light emitted from the sample. It then travels back to the dichroic mirror and through an emission and 635-nm notch filter for removal of stray incident light. Next, a cube-shaped polarizing beam splitter splits the emission into parallel (I∥) and perpendicular (I⊥) component intensities to individual linear polarizers before detection by PMTs. The fluorescein-PMT signals are converted to anisotropy via eq. 1 and by using its online fluorescence anisotropy immunoassay calibration curve, insulin concentration (thus its online secretion dynamics) can be determined.

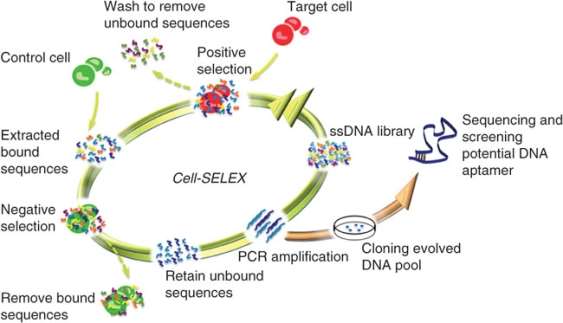
3. Created primarily by plasma cells, antibodies (or immunoglobulins) are large Y-shaped proteins recruited by the immune system to neutralize foreign agents via a precise lock-key binding mechanism. They attach to the epitope of antigens using the paratope of its fragment antigen-binding (Fab) region with high specificity. Although widely established and characterized in proteomics, antibodies have limited target potential since their targets must elicit a strong immune response for these proteins to be produced. Recently, the revolutionary development of a simple, controlled, and scalable in-vitro technique called systematic evolution of ligands by exponential enrichment (SELEX) has allowed for the isolation and identification of aptamers – which were evolved from random oligonucleotide pools. Unlike their protein counterparts, these small oligonucleotide or peptide ligands can fold into unique 3D structures than can bind to many classes of target biomolecules due their wide range in molecular recognition. Therefore, these high-affinity ligands have the benefit of unlimited diagnostic and therapeutic applications.
Aptamers can now supplement monoclonal antibodies in pharmaceutical research since they have greater advantages in their stability, in-vitro capability, size, immunogenicity, target potential, production, and ability to be modified. First, the temperature resistance of aptamers prevents denaturation and loss of structure, providing them stability at room temperature. Antibodies, contrastingly, require refrigeration as proteins denature easily, degrade over time, and have a shorter shelf life. Though, proteins have the benefit of not being affected by nuclease enzymes found in the body, which specifically cleave nucleic acid bonds. Second, aptamers are made in-vitro via SELEX; this selection means that they can be manipulated to adapt to any conditions. For antibodies, the adaptability of proteins made in-vivo are restricted to the environment of the host animal as their distribution requires the stimulation of an immune response in that live organism. Third, these smaller-sized aptamers enable them to interact with targets that may be inaccessible to the larger proteins like cell surface targets and fragments. Also, despite their improved bioavailability, they have a shortened half-life due to susceptibility to kidney filtration. Fourth, the immunogenicity of aptamers protects them from recognition by the immune system and a subsequent negative immune response. Conversely, antibodies are frequently tagged as foreign substances which, with higher dosing, increases their chances of eliciting an immune response. Fourth, with unlimited target potential, aptamers have a greater selection of targets compared to antibodies since their targets are independent of the immune system. Fifth, aptamer synthesis does not require the large-scale production of many different colonies in cell cultures that antibodies depend on – which is costly, subjected to viral or bacterial contamination, and may cause variation per batch created. Lastly, aptamers can readily adopt conjugation chemistries such as dye or functional group attachments without it being stochastic, negatively affecting activity, or leading to product mixtures – hence any shortcomings such serum degradation, variable pharmacokinetic and systemic properties, can be combated with additional modifications.
The SELEX process is an useful technique that can decipher a protein’s binding site on ss-DNA/RNA or peptides. This enrichment protocol requires the following steps: (1) define the target molecule; (2) form a large combinatorial double-stranded oligonucleotide library of DNA/RNA ligands – with primer binding sites at its ends and wobble bases in the middle for potential PCR amplification; (3) expose this pool of oligonucleotides to the target molecule; (4) partition and isolate the successful binding aptamers that have been selected by the target molecule from the non-binding ones – then amplify and subject them to additional selection cycles for further enchainment; (5) from the remaining small amount of high affinity binding molecules: isolate and sequence the individual aptamers, then refine them with altered nucleoside triphosphates like 2′-fluoro-dCTP to increase stability against endonuclease degradation by becoming unrecognized.

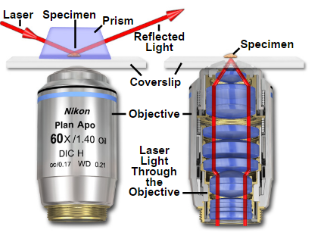
4. Total internal reflection fluorescence microscopy is a high axial-resolution imaging technique used to visualize a thin region of live specimen cells that have been incorporated with fluorescent molecules. Supported on a glass slide, this microscope optically sections the cell-substrate interface and emphasizes near membrane molecular events that are within ~100nm of the sample-coverslip contact region. Accordingly, single molecule fluorescence can be detected for fluorophores situated near adherent cell surfaces as they are being selectively irradiated, thereby minimizing excitation of fluorophores outside this focal plane and increasing the signal-to-noise ratio. This removes any out-of-focus intracellular fluorescence and reduces cellular photodamage, allowing for high-contrast and spatial resolution image production. With these advances, the biochemical kinetics and spatial-temporal dynamics of single biomolecules that are associated exclusively with process occurring at or near the plasma membrane can now be studied.
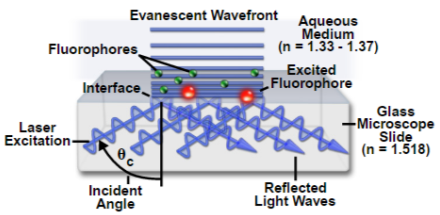
Depending on the incidence angle and refractive index differences of the two media, the collimated light beam can be reflected at the interface or refracted as it enters the second medium – limiting most of the light to the higher-index medium. In TIRFM, total internal reflection occurs as the laser excitation on the glass microscope slide (n = 1.518) propagates the light wave towards an interface of a lower-index, aqueous medium (n = 1.33-1.37) at an incident angle greater than the critical angle. The critical angle can be calculated by Snell’s Law:
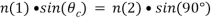 (eq.1)
(eq.1)
where n(1) is the higher refractive index; n(2) is the lower refractive index; sin(θc) is the critical incident angle relative to the normal of the interface; and sin(90) is the corresponding refracted angle. When the beam completely reflects into the microscope slide, a highly restricted electromagnetic field is induced in the specimen medium, immediately adjacent and perpendicular to the interface. With the same frequency as the incident light, this evanescent field extends a few hundred nanometers into the specimen. Since its intensity decays exponentially with distance, it can selectively excite fluorophores near the glass surface given that the energies of their electronic transitions match the wavelength bandwidth of the beam relative to its resonance conditions:
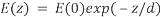 (eq.2)
(eq.2)
where E(z) is the energy at a perpendicular distance z from the interface; E(0) is the energy at the interface; (d) is the penetration depth. The secondary fluorescence emission of the fluorophores is confined to a thin region and detected by microscope optics by the prism or objective lens method.
Bowser and Khakh conducted a study to decipher the mechanisms behind astrocyte transmitter release during exocytosis by using TIRFM to image individual SpH-laden vesicles and discovered that these events were either evoked or occurred spontaneously. They used mixed hippocampal neuron-astrocyte cultures that were transfected with synaptopHluorin cDNA because this genetically encoded fluorescent SpH reporter allowed visualization of exocytosis at the single-vesicle level – by exploiting changes in pH. With the objective lens adjusted to a high numerical aperture, total internal reflection was attained with a coherent laser source and the subsequent evanescent field event excited SpHs near the coverslip-sample interface. These SpH events appeared as spontaneous increases in fluorescence intensity as the loaded vesicles became brighter when they entered the evanescent field. Eventually, as the vesicles fused with the plasma membrane, the signal rapidly decreased as the fluorescently labeled contents diffused out of the cell. In agreement with the hypothesis, ionomycin (a calcium ionophore) increased its frequency during this event, which proves that these SpH events are representative of exocytosis. For further support, the investigators compared this control group with a negative control – cells transfected with plasmids encoding for the light chain of botulinum toxin E. As expected, no SpH events were seen since this neurotoxin specifically cleaves proteins involved in synaptic vesicle exocytosis.
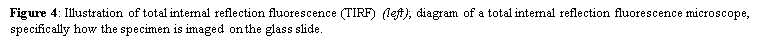
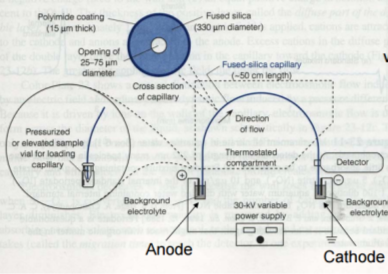
5. Gel electrophoresis is a laboratory method that uses an electric field to push a mixture of charged molecules-macromolecules or nanoparticles-through a porous (agarose or polyacrylamide) gel which serves as a separation medium. Unlike oligonucleotides, proteins and their fragments are not only separated and analyzed based on differences in size, but mostly by the magnitude of their charge. Generally, nucleic acids are sorted and visualized using agarose gel electrophoresis after being dispensed into the wells-by an operator-made during the casting of the gel. It takes advantage of DNA being negatively charged at neutral pH in the presence of ionic solutions like TAE or TBE due to its sugar-phosphate backbone. Connected to a power source, the gel is placed in an electrophoresis chamber. When an electric current is applied, they migrate from the cathode to the positively charged anode across the agarose matrix. The buffer solution serves to maintain the pH and salt concentration and contains 0.5-2.0% w/v of added agarose to successfully form a porous lattice to retard molecular motions by a sieving mechanism. Hence, shorter substrates travel faster and farther than longer ones as they can easily move through the pores, creating distinct bands based on their differential rates of migration. The gel can then be visualised with a U.V. trans-illuminator after staining the DNA with ethidium bromide. Contrarywise, capillary electrophoresis uses a fused-silica capillary tube that is filled with a polymer solution (such as hydroxyethylcellulose) instead of the traditional physical gel. It is essentially electrophoresis being conducted in a capillary tube, which also accomplishes size separation by inserting positive and negative platinum electrodes at its two ends, application of DC current and high voltage with a power supply, using buffer reservoirs for the mobile phase, and employing an on-column detector. However, the DNA samples are loaded differently by electrokinetically injecting them into the separation medium at inlet end. A positive charge is created at the outlet of the capillary to attract these negatively charged DNA via suction, which will then travel to the detector to produce a signal used to create an electropherogram. This method is the most efficient modern separation technique due to its shorter loading time and higher-resolution results, which is governed by the Van-Deemter formula where a smaller plate height indicates a higher efficiency of separation.
The velocity of solute transport down the capillary tube is governed by the following equation:
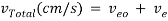 (eq. 2)
(eq. 2)
where Veo is the electroosmotic velocity; Ve is the electrophoretic velocity; and Vtotal is the apparent ionic velocity. Specifically, the electroosmotic flow of the solution describes the nature of fluid movement and occurs due to the charge distribution at the silica/capillary interface. The negatively charged, surface bound silanol groups of the fused silica (~pKa 4) contains tightly adsorbed cations – above it is the net positively charged-diffuse part of the double layer that is rich in cations. Beyond this, the bulk solution is electrically neutral. In other words, an electric double layer forms at the capillary wall. Under an electric field, the excessive solvated cations pull the water molecules during migration from the anode (inlet) towards the cathode (outlet) of the capillary, where the detector is located. This net movement of the solution front is described by the following formula:
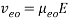 (eq. 3)
(eq. 3)
where Veo is the electroosmotic velocity; μeo isthe electroosmotic mobility; and E is the electric field strength. Moreover, the electrophoretic mobility of the solute is based on the movement of a charged molecule under an electric field, which is proportional to its charge/solute size (q/r) ratio:
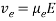 (eq. 4)
(eq. 4)
where Veo is the electrophoretic velocity; μeo isthe electrophoretic mobility; and E is the electric field strength. In the presence of electroosmotic flow, the magnitude of velocity for positive ions is greater than negative ions since they are naturally inclined to travel in the direction of the cathode rather than in reverse.
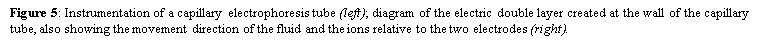

References
Question 1:
- http://ajcn.nutrition.org/content/100/3/867.short
- http://www.karger.com/Article/Pdf/345770
- http://panza.uchicago.edu/Phys.261/materials/Osmometer/
- http://onlinelibrary.wiley.com/doi/10.1111/j.1476-4431.2008.00311.x/pdf
- http://www.geminibv.nl/labware/advanced-instruments-inc.-3300-micro-osmometer/advance-micro-osmometer-3300-users-guide.pdf/view
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3886624/
- http://www.openisbn.com/preview/0471285722/%20
- http://www.iupui.edu/~cletcrse/380/ch3suppos.htm
- https://books.google.ca/books?id=z9SzvsSCHv4C&pg=PA57&lpg=PA57&dq=osmometer+instrumentation&source=bl&ots=JphQsqNWnF&sig=UJ3t-Ax6d3kHyQEjdA40I5S8wx8&hl=en&sa=X&ved=0ahUKEwjDyYqH7ZXSAhUb0IMKHSScAgcQ6AEIRjAH#v=onepage&q=osmometer%20instrumentation&f=false
- https://www.khanacademy.org/test-prep/mcat/physical-sciences-practice/physical-sciences-practice-tut/e/-using-a-freezing-point-depression-osmometer-to-measure-serum-osmolality
Question 2
- http://pubs.rsc.org/en/content/articlehtml/2017/ay/c6ay02899c
- https://www.iupac.org/publications/pac/pdf/2013/pdf/8503×0589.pdf
- http://www.horiba.com/fileadmin/uploads/Scientific/Documents/Fluorescence/Tech_Note2_-_Anisotropy.pdf
- http://link.springer.com/chapter/10.1007%2F978-0-387-46312-4_10
- https://www.picoquant.com/applications/category/life-science/fluorescence-anisotropy-polarization
- http://www.utsc.utoronto.ca/~traceslab/FLD_Anisotropy.pdf
Question 3
- http://aptamerstbc2013.wixsite.com/aptamers/vs-monoclonal-antibodies
- http://www.nature.com/nrd/journal/v9/n7/box/nrd3141_BX1.html
- https://www.researchgate.net/post/Why_is_the_monoclonal_antibody_used_more_than_the_aptamer
- http://www.basepairbio.com/research-and-publications/aptamer-applications/aptamers-antibodies/
- https://www.ncbi.nlm.nih.gov/pubmed/17627883
- https://www.google.ca/search?safe=strict&espv=2&q=advantages+of+antibodies+verus+apatmers&oq=advantages+of+antibodies+verus+apatmers&gs_l=serp.3..30i10k1.10392.13224.0.13547.15.15.0.0.0.0.155.1255.8j5.13.0….0…1c.1.64.serp..2.9.915…0i22i30k1j33i160k1j33i21k1.QfKckWwb9sI
- http://www.nature.com/nprot/journal/v5/n6/full/nprot.2010.66.html
- https://www.trilinkbiotech.com/tech/selex.asp
- https://www.ncbi.nlm.nih.gov/pubmed/21720957
Question 4
- http://www.sciencedirect.com/science/article/pii/S0091679X08006079
- https://www.ncbi.nlm.nih.gov/pubmed/11733042
- http://www.olympusmicro.com/primer/techniques/fluorescence/tirf/tirfhome.html
- http://jcs.biologists.org/content/123/21/3621.short
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2964103/
- http://www.pnas.org/content/104/10/4212.full
- https://www.microscopyu.com/techniques/fluorescence/total-internal-reflection-fluorescence-tirf-microscopy
- http://www.nature.com/nprot/journal/v1/n6/full/nprot.2006.449.html
Question 5
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal