Discovery of a Novel Oral Antithrombotic Drug Apixaban
| ✅ Paper Type: Free Essay | ✅ Subject: Chemistry |
| ✅ Wordcount: 1829 words | ✅ Published: 01 Dec 2017 |
- Shuangyu Ma
Background
Thrombosis, which is generally defined as the formation of blood clot inside blood vessels, is a major health issue in western world. Thrombosis associated disease, such as ischemic stroke and high blood pressure, remain to be the leading causes of morbidity and mortality each year in developed countries [1]. To make things worse, thrombosis is also known as a chronic disease with episodic recurrence [2]. According the American Heart Association, thirty percent of the patients diagnosed with venous thromboembolism develop recurrence within the next 10 years [2].
The balance between thrombosis formation and bleeding is prudently regulated by the blood coagulation cascade, one of the most delicate systems in human body (figure 1, [3]). The signal transmission and amplification along the cascade was conducted by a series of trypsin-like serine proteases with high specificity, often referred as zymogens [4]. The zymogens are cleaved by the previous coagulation factor to become activated proteases, which can thereby cleave the next zymogen along the cascade and achieve the signal amplification. Blood coagulation process can be triggered by intrinsic pathway or extrinsic pathway; both are followed by the activation of common pathway. The prothrombin is then cleaved by activated factor X to generate thrombin, eventually lead to the formation of stable fibrin clot.
Fig 1. Blood coagulation cascade: (figure extracted from Haematology (2nd edition) by C. J. Pallister and M. S. Watson [4])

Until now, the most well-known oral anticoagulant is warfarin, which is the current standard therapy for clinic thrombosis treatment and prevention. Despite its widely application, warfarin has a huge draw back on its dosing issue. Many of the coagulation factors, including factors II, VII, IX and X are Vitamin K dependent; the γ-carboxylation by Vitamin K is essential for their biological activity. Targeting on vitamin-K conversion cycle, warfarin produces an anticoagulation effect by reducing activities of the four vitamin K-dependent coagulation factors. However, the responses to warfarin can be interfered dramatically by genetic and environment factors, such as the common mutation on cytochrome P450 encoding gene, disease states variation, drug-drug interaction or even diet change [5]. Hence, warfarin treated patients need blood test regularly (often once a week or even worse) to determine a safe dosage case by case. Despite handling with great cautious, the anticoagulant-associated intracerebral hemorrhage keeps increasing [2]. Thus, many efforts have been put on developing a novel antithrombotic drug with low risk and high potency.
From lead to drug: the development of apixaban
The drug Apixaban, approved by FDA in 2012 for thromboembolism treatment and prevention, is developed by Pfizer and Bristol-Myers Squibb to serve as an optimized risk-benefit oral anticoagulant. The mechanism of apixaban is entirely different with warfarin – it specifically targets on activated coagulation factor ten (FXa), rather than vitamin K. Several clinical trial has proved its higher efficacy compared to asparin and lower risk compared to warfarin treatment [6].
Given the central position in the blood coagulation cascade, FXa evolved as an attractive drug target for developing anticoagulants. The DuPont Merck Pharmaceutical Company first launched a high throughput screening on FXa inhibitor in 1997 [7]. Because of the substrate similarity between FXa and the GPIIb/IIa receptor, they screened the compound library which was originally designed to be GPIIb/IIa receptor antagonist. A hit was found with low micromolar FXa affinity (coumpound 1, Ki= 38.5 μM). However, most of the reported FXa inhibitor in literature at that time posse the bisamindinoaryl structure, such as 2 and 3 (figure 2a). Inspired by these compounds, they modified the hit to be bisamidines and obtained the first list of leads (including compound 4), as shown in figure 2b.
Aiming at a higher potency, molecular modeling studies were conducted to further optimize the lead compound, which resulted in the removal of methylene unit between the isoxazoline ring and the amidine carbonyl together with the  substitution to the carbonyl. After the comparison of different substitution groups, they obtained compound 5 with Ki of 94 nM (Figure 3) [7].
Fig 2 a. The structure of hit compound and several known FXa inhibitor
 b. The bisamidine lead compounds (figure extracted from [7]).
b. The bisamidine lead compounds (figure extracted from [7]).


Fig 3. Effects of the amidine group position and  substitution (figure extracted from [7])


However, as an oral drug candidate, this dibasic lead compound might not have good oral absorption due to its hydrophilicity. Therefore, the next goal is to reduce its basicity by replacing one of the amidine groups with a neutral species. The molecular modeling of compound 5 bound to FXa indicated that the p-amidine group fits in an hydrophobic aryl-binding pocket of FXa, which is consist of three aromatic residues (Trp215, Tyr99 and Phe174, figure 4a) [8]. Therefore, it is possible to replace the p-amidine with aromatic functional groups without largely interrupt the affinity. They employed a biaryl group and obtained compound 6 with only 2-fold decrease in potency. Further investigation on different substitution at the terminal phenyl ring resulted in large enhancement on potency, such as compound 7 (Ki = 6.3 nM). (Figure 4b)
Fig 4 a. Effects of substitutions on terminal phenyl ring (figures extracted from [8]).

b. Chemical structure of compound 6 and 7

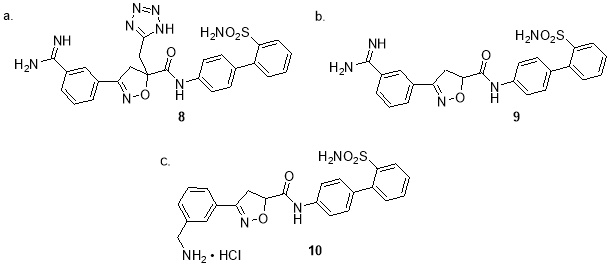
Nevertheless, another concern emerged to be the metabolic instability, as compound 7 bears an ester side chain, which is labile to esterase once it gets inside human body. Since the corresponding acid metabolite is three-fold less potent than the original ester [9], replacing the ester side chain appears becomes the imperative next step. Again, a list of compound 7 analogs bearing different side chains were synthesized and tested. Among all the functional groups, tetrazole substitution was found to be most potent, and compound 8 (Ki= 0.52 nM) was developed with additional modification on the biaryl ring motif (figure 5a). Variation on the position of amide moiety attachment and isoxazoline aromatization resulted in compound 9 (Ki= 0.15 nM, figure 5b) [10], while further investigation on a series of five-membered heterocyclic ring gives compound 10 with equal potency (Ki= 0.15 nM, figure 5c) and lower basicity [11] .
Fig 5. Chemical structure of compound 8, 9 and 10
 Despite the high potency and selectivity, compound 10 is still far from perfect due to potential mutagenicity of the biarylanilines motif [12]. Therefore, the cleavage of amide bond must be prevented by either ligating or eliminating the nitrogen (figure 6). Compound 11 was developed to have the optimized pyrazole with an even higher FXa affinity (Ki = 0.03 nM). After the scaffold was settled, more compound 11 analogs with various substitution groups and terminal aromatic rings were synthesized and compared to afford the final drug candidate as compound 12 (apixaban, Ki= 0.08, figure 7a) [1]. The optimized drug selectivity towards FXa was shown in figure 7b. The therapeutic index of the final drug compared to warfarin was demonstrated in rabbit thrombosis models and shown figure 7c) [6].
Despite the high potency and selectivity, compound 10 is still far from perfect due to potential mutagenicity of the biarylanilines motif [12]. Therefore, the cleavage of amide bond must be prevented by either ligating or eliminating the nitrogen (figure 6). Compound 11 was developed to have the optimized pyrazole with an even higher FXa affinity (Ki = 0.03 nM). After the scaffold was settled, more compound 11 analogs with various substitution groups and terminal aromatic rings were synthesized and compared to afford the final drug candidate as compound 12 (apixaban, Ki= 0.08, figure 7a) [1]. The optimized drug selectivity towards FXa was shown in figure 7b. The therapeutic index of the final drug compared to warfarin was demonstrated in rabbit thrombosis models and shown figure 7c) [6].
The drug candidate was sent for preclinical studies followed with several clinic trials, and eventually approved by FDA for venous thromboembolism prevention after hip/knee replacement and artrial fibrillation treatment. Another clinical trial for acute venous thromboembolism is still in progress.
Fig 6. Compound 11 bearing the optimized pyrazole.

Fig 7 a. Apixaban (compound 12)

b. In vitro Ki values of apixaban for FXa and other human enzymes with structural or functional similarities (figure extracted from [6]).

c. Therapeutic index of apixaban and warfarin in rabbit models (figure extracted from [6]).

Conclusion
Comparing to the initial lead compound, the eventual drug changed dramatically in chemical structure, while its FXa inhibiting potency was enhanced by 107. Tracing back to each modification step, the comprehensive consideration in drug potency, selectivity, absorption, metabolism and toxicity is truly impressive and reveals the tremendous efforts embedded in a single drug. It is worth noting that molecular modeling with FXa structure played a critical role during the rational drug modification process.
The discovery of apixaban also demonstrated the importance of defining new drug target in modern drug development. The success of apixaban is largely attributed to the better drug target FXa. However, the clinical trial of apixaban on acute coronary syndrome was discontinued due to increased bleeding events [6], which suggests that other anticoagulants are still in demand for treating various type of thrombosis.
Reference
[1] Pinto, D. J.; Orwat, M. J.; Koch, S.; Rossi, K. A.; Alexander, R. S.; Smallwood, A.; Wong, P. C.; Rendina, A. R.; Luettgen, J. M.; Knabb, R. M.; He, K.; Xin, B.; Wexler, R. R; Lam, P. Y. Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl) phenyl)-4, 5, 6, 7-tetrahydro-1 H-pyrazolo [3, 4-c] pyridine-3-carboxamide (Apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa.Journal of medicinal chemistry. 2007,50, 5339-5356.
[2] Go, A. S., Mozaffarian, D., Roger, V. L., Benjamin, E. J., Berry, J. D., Borden, W. B., Turner, M. B et al. Heart Disease and Stroke Statistics: 2013 Update: A Report From the American Heart Association. Circulation. 2013, 127, 143-146.
[3] Pallister, C. J.; Watson, M. S. Haematology, 2nd ed. Scion Publishing: Banbury, 2010; pp 336–347.
[4] Krishnaswamy, S. Exositeâ€driven substrate specificity and function in coagulation. Journal of thrombosis and haemostasis. 2005, 3, 54-67.
[5] Hirsh, J., Fuster, V., Ansell, J., & Halperin, J. L. Foundation guide to warfarin therapy. Journal of the American College of Cardiology. 2003, 41, 1633-1652.
[6] Wong, P. C.; Pinto, D. J.; Zhang, D. Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor.Journal of thrombosis and thrombolysis. 2011,31, 478-492.
[7] Quan, M. L.; Pruitt, J. R.; Ellis, C. D.; Liauw, A. Y.; Galemmo, R. A., Jr.; Stouten, P. F. W.; Wityak, J.; Knabb, R. M.; Thoolen, M. J.; Wong, P. C.; Wexler, R. R. Bisbenzamidine isoxazoline derivatives as factor Xa inhibitors.Bioorganic & Medicinal Chemistry Letters. 1997, 7, 2813-2818.
[8] Quan, M. L.; Liauw, A. Y.; Ellis, C. D.; Pruitt, J. R.; Carini, D. J.; Bostrom, L. L.; Huang, P. P.; Harrison, K.; Knabb, R. M.; Thoolen, M. J.; Wong, P. C.; Wexler, R. R. Design and Synthesis of Isoxazoline Derivatives as Factor Xa Inhibitors 1. Journal of medicinal chemistry. 1999,42, 2752-2759.
[9] Quan, M. L.; Ellis, C. D.; Liauw, A. Y.; Alexander, R. S.; Knabb, R. M.; Lam, G.; Wright, M. R.; Wong, P. C.; Wexler, R. R. Design and Synthesis of Isoxazoline Derivatives as Factor Xa Inhibitors 2. Journal of medicinal chemistry. 1999,42, 2760-2773.
[10] Pruitt, J. R., Pinto, D. J., Estrella, M. J., Bostrom, L. L., Knabb, R. M., Wong, P. C. Wright, M. R., Wexler, R. R. Isoxazolines and isoxazoles as factor Xa inhibitors.Bioorganic & medicinal chemistry letters,2000, 10, 685-689.
[11] Pinto, D. J.; Orwat, M. J.; Wang, S.; Fevig, J. M.; Quan, M. L.; Amparo, E.; Cacciola, J.; Rossi, K. A.; Alexander, R. S.; Smallwood, A. M.; Luettgen, J. M.; Liang, L.; Aungst, B. J.; Wright, M. R.; Knabb, R. M.; Wong, P. C.; Wexler, R. R.; Lam, P. Y. Discovery of 1-[3-(Aminomethyl) phenyl]-N-[3-fluoro-2′-(methylsulfonyl)-[1, 1′-biphenyl]-4-yl]-3-(trifluoromethyl) -1H-pyrazole-5- carboxamide (DPC423), a Highly Potent, Selective, and Orally Bioavailable Inhibitor of Blood Coagulation Factor Xa 1.Journal of medicinal chemistry. 2001, 44, 566-578.
[12] Pinto, D. J.; Orwat, M. J.; Quan, M. L.; Han, Q.; Galemmo, R. A., Jr.; Amparo, E.; Wwllsg, B.; Ellisj, C.; Hek, M. Y.; Alexanderf, R. S.; Knabbb, R. M.; Mersingerg, L.; Kettnera, C.; Baih, S.; Hed, K. Wexlera, R. R.; Lam, P. 1-[3-Aminobenzisoxazol-5′-yl]-3-trifluoromethyl -6-[2′-(3-(R) -hydroxy-N-pyrrolidinyl) methyl-[1, 1′]-biphen-4-yl]-1, 4, 5, 6-tetrahydropyrazolo-[3, 4-c]-pyridin -7-one (BMS-740808) a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa.Bioorganic & medicinal chemistry letters. 2006,16, 4141-4147.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal