Calcium Imaging in Neurons: Watching neurons in action
| ✅ Paper Type: Free Essay | ✅ Subject: Chemistry |
| ✅ Wordcount: 3462 words | ✅ Published: 03 Nov 2021 |
Functional imaging of neurons enables the direct visualization of neural activity at the level of action potential, calcium fluxes, intracellular signals, neurotransmitter release, and synaptic inputs. In this review, we use functional imaging to refer genetically encoded indicators of neural activity that report changes in intracellular calcium, neurotransmitter release, or voltage as changes in fluorescence. A broader definition of functional imaging might include the use of neuroimaging technology to measure an aspect of brain function and detection of neural activity by other means (for example, positron emission tomography, electroencephalography, magnetic resonance imaging, single-photon emission computed tomography or chemical dyes). Here we restrict the discussion to the general mechanism of neuronal calcium signaling, overview of the chemical fluorescent calcium indicator, and protein-based calcium indicators mostly genetically encoded calcium indicators (GECIs), Calcium imaging devices including confocal and two-photon microscopy. Finally, briefly discussing functional imaging of neurons in drosophila using GECIs as an application example to introduce new development in the field and conclude by providing an outlook on the prospects of calcium imaging.
Neuronal Calcium Signaling
Calcium (Ca2+) is an essential intracellular secondary messenger in neuron, which facilitates the neurotransmitter release in neurons, transmission of the depolarizing signal and contribute to synaptic activity (5). At rest, neuronal cells have a Ca2+ concentration of 100 nm. but are activated when this level rises to 1000 nm. (5). Two main contributing factors to cytosolic Ca2+ concentration is the equilibrium between Ca2+ influx and efflux and calcium exchange in the internal stores. Besides, the dynamics of free Ca2+ within the neuron are also determined by, calcium-binding proteins such as parvalbumin, calbindin – D28k, or calretinin, most of which serve as Ca2+ buffers (25). Multiple mechanisms cause the Ca2+ influx from extracellular space. Some of the significant contributors to neuronal calcium signaling are:
voltage-gated calcium channels (VGCC), which, based on their threshold of voltage-dependent activation, are categorized into high (HVA) and low-voltage gated channels (LVA) (9). Which group of VGCC is present in a specific neuron depends on the type of cell and the sub compartment of the cell. VGCC is effectively activated in the dendrites and spines of most central neurons through backpropagation of action potential (33) and synaptically mediated depolarization of dendritic spines (6). Since the recording of somatic Ca2+ signals are commonly used to track the action potential activity invitro (16) and invivo (30), it is essential to note that the primary determinant of these signals is VGCCs.
1) Ionotropic glutamate receptors, N-methyl-D-Aspartate receptors (NMDA) are ionotropic glutamate receptors and mediate a significant part of postsynaptic Ca2+ influx into the dendritic spines of different types of neuronal cells (35). The fraction of calcium ions that contribute to the total current via NMDA receptor channels is approximately 6% -12% (8)
2) Metabotropic Glutamate Receptors(mGluRs), this group mediates both an increase in intracellular calcium as well as a transient receptor potential type C (TRPC) channels dependent inward current (12)
3) Calcium release from internal stores, mostly the Endoplasmic reticulum (ER), regulated by triphosphate inositol receptors (IP3Rs) and ryanodine receptors (RyRS), may also include other intracellular organelles (4).
A major challenge in analyzing the different sources of neuronal Ca2+ signals is that they are not generally active at a time but overlap with strong interactions. For example, calcium influxes in the dendrites and spines of CA1 hippocampal neurons via NMDA receptors and VGCCs during strong synaptic activity are non-linear, and their combined signals serve as a coincidence detector between pre- and postsynaptic activity (35). Given these complexities, calcium imaging is often indispensable to dissect the specific mechanisms of signaling in neurons.
Calcium Indicators
Bioluminescent calcium indicator
Aequorin is a calcium-activated photoprotein isolated from the hydrozoan Aequorea Victoria (2). Bioluminescent calcium indicator aequorin is composed of the apoprotein apoaequorin and a non-covalently bound chromophore. It contains three calcium-binding sites, and upon binding of calcium ions, the protein undergoes a conformational change. The change in protein conformation results in coelenterazine oxidation to coelenteramide, resulting in photon emission (about 470 nm wavelength) due to the decline of coelenterazine from the excited state to the ground state (2). Aequorin is characterized by a high signalto-noise ratio and can monitor the concentration of cytosolic calcium between 10-7to 10-3M (2). Bioluminescent recordings of calcium signals using aequorin do not require external illumination, thereby preventing problems such as phototoxicity, photobleaching, autofluorescence, and unwanted activation of photobiological processes (27). However, each molecule performs only one cycle of emissions, and the process of recharging is relatively slow (7). Furthermore, calcium recoding based on aequorin suffers from low quantity yield to low protein stability (2).

Figure 1: Calcium Indicators (Adapted from Konnerth et al.)
Chemical Calcium Indicator
Fura-2, a representative example for the fluorescent chemical calcium indicator, is a combination of calcium chelator and fluorophore (32). Fura-2 is excitable by ultraviolet light (350/380 nm), and between 505 and 520 nm is its emission peak. Calcium ion binding causes changes in intramolecular conformation, which leads to fluorescence emitted change. Fura-2 has the advantage that it can be used with dual-wavelength excitation, with one-photon excitation. Thus fura-2 enables the quantitative determination of the calcium concentration in a neuron of interest to be independent of the concentration of intracellular dye (32). Another important advantage of chemical calcium indicator is that they exist in a membrane-permeable and membraneimpermeable form, allowing their use in combination with a variety of different loading techniques (32). A significant drawback is that the cellular localization of chemical Ca2+ indicators cannot be easily controlled or explicitly targeted to a specific organelle (14).
Genetically encoded calcium indicators (GECIs)
GECIs are available in two flavors, those involving Forster resonance energy transfer (FRET) and single fluorophore. Here we have chosen Yellow Cameleon (YC) as a representative to display the GECIs based on FRET (18). FRET is the phenomenon on which the Cameleon sensor family relies. It occurs between closely applied fluorophores (donor and acceptor) that overlap the spectrum of emissions and excitation (17). The extent of FRET depends on the degree of overlap between the two spectra, the fluorescence dipole orientation, and, crucially, the distance between the two spectra. The degree of overlap between two spectra is very responsive to the distance between fluorophores over the 1-10 nm scale (17). YC 3.60 is made up of two fluorescent proteins and belongs to the GECI Cameleon family (17). YC consists of the enhanced cyan fluorescent-cent protein (ECFP) as a donor and the circularly permuted Venus protein as an acceptor, a linker sequence composed of calcium-binding protein calmodulin, and calmodulin-binding peptide M13 binds these two proteins (17). In the absence of calcium ions, the blue ECFP fluorescence (480nm) dominates the emission. Upon calcium binding, intramolecular conformation changes lead to reduction, which refers to a form of non-radiative energy transfer between an excited fluorophore donor and a fluorophore acceptor. Thus, due to the occurrence of FRET, the Venus protein is excited and emits photons (530 nm). The blue fluorescence is declining in practice, while the yellow fluorescence is increasing. The calcium signal is represented as the Venus-ECFP fluorescence ratio. To stop potential associations with endogenous binding partners of calmodulin, two specific methods have been introduced. The calmodulin-M13-binding interfaces are mutated in D3cpV-type GECIs to minimize cellular target interactions (17) substantially. Calmodulin is replaced by troponin C variants in another type of FRET-based calcium indicators. Troponin C is the calcium-binding protein in the cells of the cardiac and skeletal muscles and, as such, has no endogenic binding neuron partners (15).
The archetypal GECI is GCaMP, is a genetically encoded calcium marker, GCaMP is produced from a fusion of green fluorescent protein (GFP) circularly permuted (i.e., provided new N- and Ctermini) and M13, a peptide sequence from myosin light chain kinase and fused to calcium-binding protein calmodulin; The structural rearrangement of the sensor removes the GFP barrel in the presence of calcium, which dramatically increases the fluorescence output (19 ) . The original version has been improvised and reduplicated (by multiple groups) through rational design and selective mutagenesis to increase the total fluorescence change in response to calcium, regulate calcium-binding affinity, and accelerate response time (kinetics)— resulting in the recent generation of jGCaMP7(10).
Dye Loading Approaches
The loading of calcium indicators into neurons would rely mainly on three factors, the form of calcium indicator, the biological preparation, and the accurate scientific topic being discussed. The three most commonly employed approaches for dye loading of individual neuron are:
a) Single-cell loading by hard microelectrode impalement, whole-cell patch-clamp mounting, and single-cell electroporation, which can be used to load chemical and genetically encoded calcium markers (23,32).
b) Acute network loading. Most neurons are marked by acetoxymethyl ester (AM), by dextranconjugated dye c and by bulk electroporation simultaneously (23,32).
c) Genetically encoded calcium indicators (GECI) are transmitted via viral transduction, in utero electroporation, and by generation of transgenic lines (23,32).
Common Calcium Imaging Devices
The main types of instruments used for calcium imaging are:
Widefield microscopy
Using a Photodiode array or a or a charged coupled device (CCD)-based camera. The light source is typically a mercury or xenon lamp in such cases, making a simple adjustment of the wavelengths of the excitation. Spin between two wavelengths of excitation, as seen in ratiometric measures of excitation, can be performed by using either a filter wheel or a monochromatic light source. Excitation and emission light usually are distinguished by a dichroic mirror located within the microscope. The photodiode arrays can be used to carry out calcium imaging. The traditional photodiode array comprises a series of photodiode (typically 124-1020 elements). Every photodiode is one pixel. Photodiode arrays are characterized by very high dynamic range and high speed but rather low spatial resolution. CCD-based cameras consist of many photodiodes densely packed on a chip with a serial readout of the signals. The newest generation of CCD-based cameras has an exquisitely high spatial and temporal resolution, but in some cameras, the noise level per pixel is high. The contrast and resolution of calcium imaging based on wide-field microscopy is limited by light dispersion, especially when attempting to image neurons more profound in the brain tissue (22,30). Such limitations make it more suitable for in vitro neuronal calcium imaging, like calcium imaging in neuronal cell cultures (30)
Laser scanning microscopy
Calcium imaging of neurons at deeper brain locations is usually done with confocal or two-photon microscopy. Using a laser beam in a raster pattern specimen is scanned. The fluorescent light emitted from each scan point (fluorescence value acquired for each pixel) is captured by a detector to create an image. Confocal microscopy, as a light source, uses a continuous wave laser. A scanner monitors the excitation position Confocal system across the sample.

Figure 2: Scanning Confocal and spinning disk
Upon passing a pinhole that blocks out-of-focus fluorescence, the emission light is detected and enters the photomultiplier tube. A dichroic mirror distinguishes the excitation and emission light. A significant limitation is that confocal aperture often blocks photons that are generated in the focal plane but are distributed on the way back through the optical path. Confocal microscopy, like wide-field microscopy, is therefore often limited to in vitro preparations, such as cultured neurons or brain slices. Eventually, some applications benefit from the use of disc-based confocal imagery, which requires the use of a spinning disc with many fine pinholes, each of which functions as an individual confocal aperture. A CCD -based camera can be used to detect images. Due to simultaneous sampling from several focal points, the spinning disc confocal approach can achieve higher image acquisition rates than laser scanning confocal microscopes (14.,34). Figure 2 provides a schematic image of the confocal microscope and the spinning discs confocal microscope.
A significant step forward in the field of neuroscience was the development of two-photon microscopy that enables high-resolution and high-sensitivity fluorescence microscopy in widespread in vivo brain tissue (20,29). Through two-photon microscopy, two low-energy near-IR photons come together to create a transition from the ground to the excited state in a fluorescent molecule. Figure 3, schematic image of the twophoton microscope The advantage is that the usual wavelengths of excitation are within the near-IR spectrum, with better tissue penetration than the visible light used in single-photon microscopy(20).It makes measurements of fluorescence concerning neuronal or vascular brain activity at > 100 micrometers deeper than standard objectives(20,31 ).
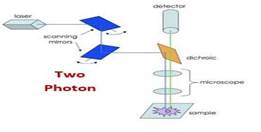
Figure 3: Two-Photon microscopy
Another significant advantage is that the fluorescence background frequency is shallow. For all these reasons, two-photon calcium imaging has become the preferred method of monitoring in deeper brain regions. Cortical circuits with well-preserved connections can be studied in vivo. Standard twophoton lasers can be tuned from 700 nm to 1000 nm or more and can be used to excite most commercially available fluorophores (20,29,31).
Functional imaging of neurons employing GECI in Drosophila
While cellular calcium transients are slower than the underlying electrical activity, all variants of GCaMP respond over relevant time scales to detect neural activity in fly neurons. GECIs have been optimized for chemical dyes that are sensitive to voltage or calcium (28). Comparisons between GCaMP responses and simultaneous electrophysiological stimulation or fly neuromuscular junction (NMJ) and olfactory lobe neurons demonstrate that fluorescence variations in GCaMP consistently represent a decrease and increases in neural activity in the neuronal populations in which it is expressed. Under certain conditions and in some neurons, GCaMP6 is even capable of monitoring single action potentials in single tests in live drosophila larvae (11), and jGCaMP7 sensors were shown to image a population of compass neurons in adult drosophila during behavior (10). Synaptically targeted calcium indicators have also been used to compare the activity levels of the different fly NMJ buttons (28). The generic fluorescence protein can be fused directly to the GCaMP, for example, with the Green-OrangeMatryoshka-GCaMP6 fusion “nested doll” (1), CaMPARI (11), and Photocon- vertible calcium indicators (28) enable user-specific classification of specific cells of interest, e.g., in areas where neuron populations are intertwined. GECIs are not promising “micro-scope electrophysiology,” but are new tools with their applications. Experiments requiring electrophysiological accuracy with GECIs should not be attempted realistically. GECIs enable a different type of experiment to visualize neural activity in many recognizable neurons simultaneously (3,10,11). GECIs are well suited to classify brain areas with corresponding activity, map regions that react to stimuli in more intact and behavioral animals, agnostically monitor for linked brain areas, and see how activity is progressing across sequential layers in the neural circuit. Several calcium sources may cause changes in cellular calcium levels. As GCaMP records calcium-binding, regardless of the origin of calcium, alternative sources should always be considered. Adding a voltage-gated calcium channel blocker or an actionpotential blocker could be used to verify that most of the fluorescence shift recorded by the GECI was due to action potential related influx of Ca2+ (28).
Imaging analysis
Typical usable image data is obtained as movies or Z-stacks in software supplied by microscope vendors. These files are often large and, by selecting precise regions of interest, files can be compressed, downsampled, or reduced. Analytics software packages are available on the ImageJ / FIJI platform (24) or custom scripts written by individual lab in MATLAB, R, or Python. Data is usually reported as a fluorescence shift, divided by the baseline (y-axis) versus the time (x-axis), with an image of the interest area where the signal is measured.
Fly line recommendations
There are numerous GECI fly lines available at the Bloomington Drosophila Stock Center (http://fly.bio.indiana.edu) infused with the newest generation of GCaMPs for imaging in presynaptic terminals. Many laboratories produce and optimize genetically encoded indicators for use in flies; one site that is recommended for use in fly’s potential users check is the Howard Hughes Medical Institute Janelia Genetically Encoded Neuronal Indicator and Effector (GENIE) Project site (https://www.janelia.org/project-team/genie/tools).
Conclusion
New generation of calcium indicators are being developed and published at a brisk pace, with each new generation offering brightness, photostability, sensitivity, and kinetics improvements. Specialized GECIs that could provide a better resolution of action potential acts and the timing of fast-spiking neurons. GECIs for the entire complement of neurotransmitters, neuromodulators, and secondary messengers are being created, potentially allowing complete investigation of molecular signaling in the neural circuit. What are the next significant challenges in calcium neuronal imaging? Another critical area that is likely to expand sharply in the coming years is calcium imaging in specified types of neurons awake, animals behaving. Such experiments are not limited to drosophila, c. elegans, but are likely to be applied to other types as well, such as ferrets, dogs, and primates. The use of calcium imaging in molecular medicine for a detailed analysis of signaling pathways in the explosively increasing number of disease models, particularly neurodegenerative diseases like Alzheimer’s and Parkinson’s. Lastly, calcium imaging in neurons can benefit significantly from the improved GECIs with higher signal sensitivity and better characteristics of transient response, and times were never better for quick, efficient circuit mapping in any model organism.
References
1. Ast C, Foret J, Oltrogge LM, et al. Ratiometric Matryoshka biosensors from a nested cassette of green- and orange-emitting fluorescent proteins. Nature Communications. 2017;8.
2. Bakayan A, Vaquero CF, Picazo F, Llopis J. Red Fluorescent Protein-Aequorin Fusions as Improved Bioluminescent Ca2+ Reporters in Single Cells and Mice. Plos One. 2011;6(5).
3. Berlin K, Koren S, Chin CS, Drake JP, Landolin JM, Phillippy AM. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol. 2015;33(6):623-630.
4. Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21(1):13-26.
5. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nature Reviews Molecular Cell Biology. 2000;1(1):11-21.
6. Bloodgood BL, Sabatini BL. Ca2+ signaling in dendritic spines. Current Opinion in Neurobiology. 2007;17(3):345-351.
7. Brini M. Calcium-sensitive photoproteins. Methods. 2008;46(3):160-166.
8. Burnashev N, Zhou Z, Neher E, Sakmann B. FRACTIONAL CALCIUM CURRENTS THROUGH
RECOMBINANT GLUR CHANNELS OF THE NMDA, AMPA AND KAINATE RECEPTOR
SUBTYPES. Journal of Physiology-London. 1995;485(2):403-418.
9. Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annual Review of Cell and Developmental Biology. 2000;16:521-555.
10. Dana H, Sun Y, Mohar B, et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nature Methods. 2019;16(7):649-+.
11. Fosque BF, Sun Y, Dana H, et al. Neural circuits. Labeling of active neural circuits in vivo with designed calcium integrators. Science. 2015;347(6223):755-760.
12. Hartmann J, Dragicevic E, Adelsberger H, et al. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59(3):392-398.
13. Helmchen F, Waters J. Ca2+ imaging in the mammalian brain in vivo. European Journal of Pharmacology. 2002;447(2-3):119-129.
14. Lichtman JW, Conchello JA. Fluorescence microscopy. Nature Methods. 2005;2(12):910-919.
15. Mank M, Santos AF, Direnberger S, et al. A genetically encoded calcium indicator for chronic in vivo two-photon imaging. Nat Methods. 2008;5(9):805-811.
16. Mao BQ, Hamzei-Sichani F, Aronov D, Froemke RC, Yuste R. Dynamics of spontaneous activity in neocortical slices. Neuron. 2001;32(5):883-898.
17. Miyawaki A, Griesbeck O, Heim R, Tsien RY. Dynamic and quantitative Ca2+ measurements using improved cameleons. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(5):2135-2140.
18. Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci U S A. 2004;101(29):10554-10559.
19. Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol. 2001;19(2):137-141.
20. Oheim M, Beaurepaire E, Chaigneau E, Mertz J, Charpak S. Two-photon microscopy in brain tissue:
parameters influencing the imaging depth. J Neurosci Methods. 2001;111(1):29-37.
21. Ohmiya Y, Hirano T. Shining the light: the mechanism of the bioluminescence reaction of calciumbinding photoproteins. Chem Biol. 1996;3(5):337-347.
22. Ratzlaff EH, Grinvald A. A TANDEM-LENS EPIFLUORESCENCE MACROSCOPE – HUNDRED-FOLD BRIGHTNESS ADVANTAGE FOR WIDE-FIELD IMAGING. Journal of
Neuroscience Methods. 1991;36(2-3):127-137.
23. Rehberg M, Lepier A, Solchenberger B, Osten P, Blum R. A new non-disruptive strategy to target calcium indicator dyes to the endoplasmic reticulum. Cell Calcium. 2008;44(4):386-399.
24. Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676-682.
25. Schwaller B. Cytosolic Ca2+ Buffers. Cold Spring Harbor Perspectives in Biology. 2010;2(11).
26. Segal M. IMAGING OF CALCIUM VARIATIONS IN LIVING DENDRITIC SPINES OF CULTURED RAT HIPPOCAMPAL-NEURONS. Journal of Physiology-London. 1995;486(2):283-
295.
27. SHIMOMURA O, JOHNSON FH, SAIGA Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J Cell Comp Physiol. 1962;59:223-239.
28. Simpson JH, Looger LL. Functional Imaging and Optogenetics in Drosophila. Genetics. 2018;208(4):1291-1309.
29. Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(12):7319-7324.
30. Svoboda K, Denk W, Kleinfeld D, Tank DW. In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature. 1997;385(6612):161-165.
31. Svoboda K, Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 2006;50(6):823-839.
32. Tsien RY, Rink TJ, Poenie M. MEASUREMENT OF CYTOSOLIC FREE CA-2+ IN INDIVIDUAL SMALL CELLS USING FLUORESCENCE MICROSCOPY WITH DUAL EXCITATION WAVELENGTHS. Cell Calcium. 1985;6(1-2):145-157.
33. Waters J, Schaefer A, Sakmann B. Backpropagating action potentials in neurones: measurement, mechanisms and potential functions. Prog Biophys Mol Biol. 2005;87(1):145-170.
34. Xu C, Zipfel W, Shear JB, Williams RM, Webb WW. Multiphoton fluorescence excitation: New spectral windows for biological nonlinear microscopy. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(20):10763-10768.
35. Yuste R, Denk W. DENDRITIC SPINES AS BASIC FUNCTIONAL UNITS OF NEURONAL INTEGRATION. Nature. 1995;375(6533):682-684.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal