Acute Myeloid Leukemia: Causes and Treatment Strategies
| ✅ Paper Type: Free Essay | ✅ Subject: Chemistry |
| ✅ Wordcount: 2174 words | ✅ Published: 30 Jan 2018 |
Pathology
Acute myeloid leukemia is a disease that primarily afflicts adults. The likelihood of being diagnosed with AML increases with age; the median age of diagnosis is 65 with very few cases reported in those under the age of 40 [4]. Several risk factors have been associated with increased incidence of AML including: Li-Fraumeni disease, Klinefelter’s syndrome, radiation exposure, chemotherapy, and chemicals (benzene, herbicides, etc) (See supplementary figure S.1) [4]. However, the initiation of AML is a multistep process and can be the result many different genetic aberrations [4][5]. Therefore, the aforementioned risk factors do not account for all cases of AML [4].
Acute myelogenous leukemia is the result of oncogene-driven accumulation of immature myeloblasts within the bone marrow [5]. Myeloblasts are progenitor cells, which will ultimately give rise to neutrophils, basophils, eosinophils, and mast cells (collectively known as the granulocytes) [6]. In AML, the genes that govern proper differentiation of myeloblasts into one of the aforementioned cell types are mutated. This prevents differentiation and leads to a buildup of myeloblasts within the marrow [5]. The clinical consequences of myeloblast buildup are marrow failure leading to low white-blood cell count, low red-blood cell count, and insufficient levels of clotting factors [5]. Therefore, clinical symptoms are depressed immune function, anemia, and continued hemorrhaging.
The molecular pathogenesis of AML (and all cancers) begins with the acquisition of genetic abnormalities. There are two models that describe how these acquisitions arise. The conventional model of cancer cell initiation proposes that the cell gradually acquires certain mutations to genes involved in mitotic signaling (KRAS or APC) and tumor suppression (P53), thereby allowing the cell to divide uncontrollably [7]. However, Recent work by Stephens et al. [8] showed that multiple mutations can be induced in a “one off” event by the random shattering and re-ligation of one or more chromosomes [8]. They termed this event “Chromothripsis” [8]. Chromothripsis results in massive translocations and changes to copy number state, but is distinct from the conventional model of cancer cell initiation by the presence of large-scale inter-chromosomal rearrangements [8]. Thus, the chromothripsis model differs from its conventional counterpart in the time taken for the cell to reach malignancy and the scale to which the genome is altered.
Chromothripsis is observed in a portion of AML cases; Rausch et al. [9] found nearly half of the AML cohort showed chromosomal rearrangements consistent with chromothripsis [9]. This shows that, accumulation of the necessary mutations that drive AML may occur by more than one mechanism.
By either mechanism of AML initiation, myeloblasts lose the ability to differentiate. The molecular pathogenesis commonly shows two frequent chromosomal aberrations – a translocation between chromosome 8 and 21, and an inversion of chromosome 16 [5]. These changes affect two genes crucial for myeloid differentiation (CBF1α and CBF1β) [5]. The effect of the inversion and translocation results in a gene chimera, which is translated into a protein that interferes with proper CBF1α and CBF1β function [5]. However, these specific chromosomal alterations are not observed in every case of AML. DNA-damage inducing agents like radiation or certain chemicals, may cause aberrations to chromosome 5 and 7, which has also been implicated in the initiation of AML [5]. This shows that there are several factors involved in proper myeloid differentiation and that interference to any of them may result in AML.
Visual differentiation of healthy myeloblasts from leukemic myeloblasts can be challenging. Myeloblasts should contain 3 – 5 nucleoli, which are full of uncondensed chromatin [6]. Some leukemic myeloblasts may show more than 3 – 5 nucleoli [5]. Also, they do not normally contain granules, however, leukemic myeloblasts may have granules, which can serve as a potential marker for diagnosis [5]. It is important to stress that these morphological changes may not appear in all cases of AML. Therefore, prognosis is confirmed by the presence of greater than 20% myeloblasts in bone marrow biopsy [5].
Treatment
There are different avenues for treating AML. Treatment may include supportive care (in advanced cases), chemotherapy, and stem cell transplantation. However, chemotherapy is the most common and effective method of treatment [3]. When medicinal chemists began isolating antibiotics produced from bacteria in search of potential leads for drug design, Aurelio Di Marco and his research team discovered a new species of bacteria, Streptomyces peucetius, within a soil sample they obtained from an area near Bari, Italy [10][11]. This new strain of bacteria was produced a compound that was efficacious as a chemotherapeutic agent against many tumors; the compound was later named daunorubicin and is now considered a key intervention administered to patients with AML [12]. Daunorubicin and doxorubicin are part of the class of antibiotics collectively known as the anthracyclines.
 Anthracyclines can cause cytotoxicity by different mechanisms depending on their intracellular concentration. As reviewed by Gerwitz [13], in vitro studies show there are several possible mechanisms for the anti-tumor effects of these agents including: Inhibition of DNA synthesis, free radical generation leading to either DNA damage or lipid peroxidation, inhibition of DNA topoisomerase resulting in helix super-coiling, DNA alkylation, and DNA cross-linking [13]. The induction of apoptosis was also mentioned as a mechanism of cytotoxicity, but it is likely that apoptosis is a byproduct of the aforementioned cellular stresses, rather than a direct consequence of anthracycline exposure [13]. It is important to note that these mechanisms were observed in vitro, and that in vitro conditions allow for exposure at concentrations that may greatly exceed in vivo concentrations [13].
Anthracyclines can cause cytotoxicity by different mechanisms depending on their intracellular concentration. As reviewed by Gerwitz [13], in vitro studies show there are several possible mechanisms for the anti-tumor effects of these agents including: Inhibition of DNA synthesis, free radical generation leading to either DNA damage or lipid peroxidation, inhibition of DNA topoisomerase resulting in helix super-coiling, DNA alkylation, and DNA cross-linking [13]. The induction of apoptosis was also mentioned as a mechanism of cytotoxicity, but it is likely that apoptosis is a byproduct of the aforementioned cellular stresses, rather than a direct consequence of anthracycline exposure [13]. It is important to note that these mechanisms were observed in vitro, and that in vitro conditions allow for exposure at concentrations that may greatly exceed in vivo concentrations [13].
Pharmacodynamics
 Although, daunorubicin and doxorubicin are thought to cause cytotoxicity by several different processes, their ability to bind to DNA and prevent DNA replication or transcription is considered to be the primary means of anti-tumor activity in vivo [13][14]. This is because replication is inhibited at anthracycline concentrations that can be reached in vivo following a standard dosage [13]. The two anthracyclines appear to intercalate preferentially to regions of DNA with select base-pair composition, specifically, regions with CGATCG sequences [14]. Rabanni, Finn, & Ausio [15] suggest that intercalation stabilizes hydrogen bonds between the two strands of DNA [15]. This means that the energy required to separate the strands is higher, and cannot be achieved by DNA helicase [15]. Thus, strand separation is inhibited, preventing DNA replication and tumor cell proliferation.
Although, daunorubicin and doxorubicin are thought to cause cytotoxicity by several different processes, their ability to bind to DNA and prevent DNA replication or transcription is considered to be the primary means of anti-tumor activity in vivo [13][14]. This is because replication is inhibited at anthracycline concentrations that can be reached in vivo following a standard dosage [13]. The two anthracyclines appear to intercalate preferentially to regions of DNA with select base-pair composition, specifically, regions with CGATCG sequences [14]. Rabanni, Finn, & Ausio [15] suggest that intercalation stabilizes hydrogen bonds between the two strands of DNA [15]. This means that the energy required to separate the strands is higher, and cannot be achieved by DNA helicase [15]. Thus, strand separation is inhibited, preventing DNA replication and tumor cell proliferation.
For both compounds, the chromophore portion (DCBA in fig. 1) of the molecule intercalates between complementary guanine-cytosine (GC) base pairs on each side of the DNA double-helix (fig. 2). The compound is stabilized in this position by hydrogen bonds on either side of the molecule. The “D” side of the molecule is held in position with aid from a solvent atom (believed to be ionic sodium), which forms hydrogen bonds between oxygen 4 and 5 to nitrogen 7 of guanine12 (fig. 2) [14]. The “A” side of either drug is coordinated by hydrogen bonds from oxygen 9 to nitrogens 2 and 3 of guanine 2 (fig. 2) [14]. As previously mentioned, all of these interactions occur between both daunorubicin/doxorubicin and C/G DNA sequences. However, the added hydroxyl group on C14 of doxorubicin creates an extra association. The hydroxyl appears to shift solvent molecules such that indirect interactions between itself and the phosphate backbone of DNA are created (fig. 3) [14]. This added interaction might explain the slightly higher in vitro affinity constant between doxorubicin-DNA (Ka = 1.6×10-5) as compared to daunorubicin-DNA (Ka = 1.2×10-5) (in vitro Ka determination performed at 37°C in 10% fetal calf serum) [17].
 Doxorubicin is more potent than daunorubicin. In the case of anthracycline induced inhibition of DNA strand separation, the potency and efficacy are directly related. The increased binding affinity of doxorubicin means that it will associate with DNA and prevent DNA separation at a lower concentration (See supplementary fig.S2). Therefore, pharmacological factors aside, it will be more potent and will prevent tumor cell proliferation at lower concentrations.
Doxorubicin is more potent than daunorubicin. In the case of anthracycline induced inhibition of DNA strand separation, the potency and efficacy are directly related. The increased binding affinity of doxorubicin means that it will associate with DNA and prevent DNA separation at a lower concentration (See supplementary fig.S2). Therefore, pharmacological factors aside, it will be more potent and will prevent tumor cell proliferation at lower concentrations.
Synthesis
Doxorubicin is a semi-synthetic analog of daunorubicin. The daunorubicin producing bacteria, Streptomyces peucetius, was mutated to the doxorubicin producing subspecies Streptomyces peucetius subsp. caesius [18]. This mutated strain was found to carry an oxidizing cytochrome P450 isoform capable of hydroxylating the C14 of daunorubicin [19]. The gene that encodes this P450 is called doxA [19]. Although, this gene is crucial for doxorubicin production, its presence does not guarantee collectable quantities of doxorubicin (See supplemental figure S.2) [20]. Enzymes, such as dnrH and dnrX, may further oxidize or reduce the newly formed doxorubicin into other metabolites that do not have antitumor abilities [18]. Therefore, in order to make doxorubicin production sustainable, the down regulation of such genes is required to increase doxorubicin yield.
Pharmacokinetics
Despite their similar molecular structure, daunorubicin and doxorubicin have distinct pharmacokinetic properties. This section will begin by contrasting the differences in elimination, metabolism, and distribution. Bioavailability will not be contrasted because both of these agents are administered intravenously. The section will then conclude with a description of tissue distribution, intracellular distribution, and toxicity.
Administration
As already mentioned, both anthracyclines are administered via intravenous injection [21][22]. They should not be injected subcutaneously or intramuscularly due to the significant risk of tissue death at and near the injection site [23]. The exact dosage schedule for both daunorubicin and doxorubicin is dependent on several different factors including: age, health, pre-exisiting heart conditions, and renal functioning, therefore exact dosages vary among different patient populations. In terms of daunorubicin, dosage ranges between 30 and 45 mg/m2, whereas doxorubicin dosages range between 40 and 75 mg/m2 during the initial course of treatment [24][25].
Elimination

 Both compounds are eliminated from the plasma via urinary and biliary mechanisms [23]. However, a much greater proportion of a daunorubicin dose is excreted through the kidneys (14-23%) as compared to doxorubicin (5%), while a
Both compounds are eliminated from the plasma via urinary and biliary mechanisms [23]. However, a much greater proportion of a daunorubicin dose is excreted through the kidneys (14-23%) as compared to doxorubicin (5%), while a
larger proportion of doxorubicin was reported to be excreted through the bowels (50%) compared to daunorubicin (40%) [23].
Daunorubicin and doxorubicin are considered to be high clearance drugs. They have a similar clearance value of 1-2 L/Kgï‚Ÿh [26]. The elimination of either drug, along with their metabolites, follows a triphasic decline in plasma concentration [23]. After a 60ml/m2 infusion of daunorubicin/doxorubicin, the plasma concentration/time graph shows that both drugs are eliminated with a similar rate (fig.3) [26]. However the Medsafe® [23] datasheet for daunorubicin/doxorubicin reports that the plasma T1/2 for the first phase averages 45min daunorubicin and 12min doxorubicin, while the second phase is 18.5hrs daunorubicin, 3.3hrs doxorubicin.
Metabolism
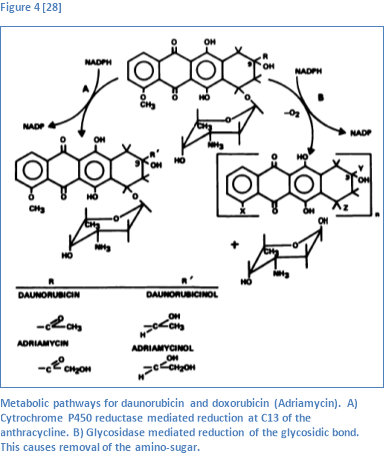 The primary metabolites of daunorubicin and doxorubicin are daunorubicinol and doxorubicinol respectively. Because of their similar structures, the metabolism of either compound occurs in a likewise fashion. The enzyme most implicated in metabolism is NADPH (nicotinamide-adenine-dinucleotide-phosphate) dependent cytochrome P450 reductase [27]. This enzyme reduces the ketone at C13 to an alcohol to produce daunorubicinol and doxorubicinol [28]. These two products are believed to retain their cytotoxicity and are cleared from the plasma at a similar rate [28][29].
The primary metabolites of daunorubicin and doxorubicin are daunorubicinol and doxorubicinol respectively. Because of their similar structures, the metabolism of either compound occurs in a likewise fashion. The enzyme most implicated in metabolism is NADPH (nicotinamide-adenine-dinucleotide-phosphate) dependent cytochrome P450 reductase [27]. This enzyme reduces the ketone at C13 to an alcohol to produce daunorubicinol and doxorubicinol [28]. These two products are believed to retain their cytotoxicity and are cleared from the plasma at a similar rate [28][29].
Another common metabolite for either compound is generated via reduction of the glycosidic bond by gycosidases. Glycosidic reduction results in removal of the amino-sugar that is bonded to C7 [28]. This reaction eliminates the anti-tumor capabilities of either compound [28]. Following removal of the amino-sugar, the remaining aglycone is demethylated and then undergoes phase II metabolism (sulfonation or glucuronidation) [23]. Phase II metabolism by sulfonation or glucuronidation increases plasma clearance and enhances elimination. Two other metabolites are listed in literature, 7-deoxydoxorubincolone and 7-deoxydoxorubinone, however the enzyme or enzymes that facilitate these reactions were not noted [30].
Two other enzymes metabolize both daunorubicin and doxorubicin under aerobic conditions, xanthine dehydrogenase and xanthine oxidase, and produce reduced semiquinones and oxygen radicals [31]. These radicals could provide added efficacy to the drug therapy. It has been suggested that advanced stage cancer cells exhibit high oxidative stress and that it may be advantageous to exploit this finding to preferentially and specifically attack and destroy them [30]. Under anaerobic conditions, xanthine dehydrogenase metabolizes doxorubicin to 7- deoxydoxorubicin aglycone, which has been found to be efficacious in the reduction of the drug’s toxicity [31].
Volume of Distribution
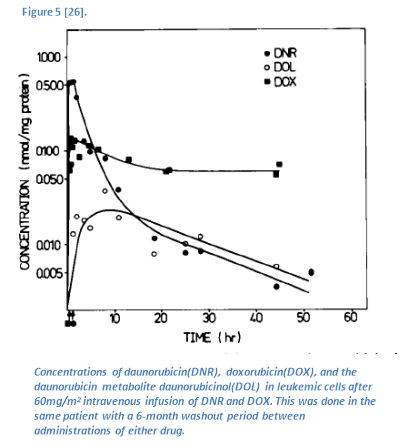 Daunorubicin and doxorubicin localize in the tissues. The Vd for daunorubicin is 39.2 L/Kg, while the Vd for doxorubicin is 24.6 L/Kg [26]. This indicates that both compounds have high tissue distribution [26]. Although the addition of a hydroxyl group at C14 markedly reduces doxorubicin’s ability to distribute into the tissues (relative to daunorubicin), doxorubicin remains within leukemic cells for a longer duration than daunorubicin (fig. 5) [26]. Upon administration, daunorubicin accumulates within leukemic cells more rapidly than does doxorubicin (fig 5). This is likely because of the increased polar surface area brought on by the added hydroxyl group of doxorubicin, which hinders its ability to cross membranes. However, the added hydroxyl group provides increased binding affinity to DNA [17]. Therefore, it will not leave the cell as rapidly because it will remain bound to DNA [26].
Daunorubicin and doxorubicin localize in the tissues. The Vd for daunorubicin is 39.2 L/Kg, while the Vd for doxorubicin is 24.6 L/Kg [26]. This indicates that both compounds have high tissue distribution [26]. Although the addition of a hydroxyl group at C14 markedly reduces doxorubicin’s ability to distribute into the tissues (relative to daunorubicin), doxorubicin remains within leukemic cells for a longer duration than daunorubicin (fig. 5) [26]. Upon administration, daunorubicin accumulates within leukemic cells more rapidly than does doxorubicin (fig 5). This is likely because of the increased polar surface area brought on by the added hydroxyl group of doxorubicin, which hinders its ability to cross membranes. However, the added hydroxyl group provides increased binding affinity to DNA [17]. Therefore, it will not leave the cell as rapidly because it will remain bound to DNA [26].
Toxicity
The clinical use of doxorubicin and daunorubicin may be limited by their similar inherent toxicity. It goes without saying that chemotherapeutics should not be administered to women who are lactating or pregnant due to significant health complications that can be incurred by the infant or fetus, respectively, therefore these drugs should not be considered in these populations [33]. The use of these agents in a long-term manner is cautioned due to their ability to cause cardiotoxicity [29]. Under aerobic conditions, the semiquinone radical that may be produced during metabolism undergoes redox cycling [27]. Accumulation of reaction oxygen species soon occurs and can cause cytotoxicity [27]. This is believed to account for tissue death in cardiac cells, and is one of the major implications of using the anthracycline class of anti-tumors agents [27]. The cardiotoxicity itself includes effects such as functional damage to mitochrondrial DNA, interference of cytochrome c oxidase, and dysfunctional lipid peroxidation [13].
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal