Role of Exercise in Reducing Insulin Resistance
| ✅ Paper Type: Free Essay | ✅ Subject: Biology |
| ✅ Wordcount: 2574 words | ✅ Published: 06 Jun 2018 |
- By: Pranav Maddali
Abstract
The major pathway of glucose regulation in the body is via the Glucose Transport System (GTS) in which cells stimulated by insulin cause an upregulation in expression and translocation of a glucose transporter protein, GLUT4, to the cell plasma membrane, resulting in increased glucose transport into the cell. Thus, insulin is directly responsible for regulating carbohydrate and fat metabolism in the body. However, increased dietary intake and the lack of physical exercise has been implicated in creating a condition known as “insulin resistance”, whereby the cells fail to uptake glucose in response to insulin stimulation, leading to hyperglycemia and significantly increasing the odds of acquiring insulin-dependent (Type-1) and non-insulin-dependent diabetes mellitus (Type-2). However, recent studies have shown that skeletal muscles, the major consumer of glucose in the body, possess an alternative mechanism to elicit glucose uptake using GLUT4 instead of insulin stimulation, which is triggered by exercise. This review elucidates glucose transport mechanisms in both normal and insulin resistance states and the role of physical exercise in potentially reversing insulin resistance and helping regulate blood glucose levels in diabetic patients.
INTRODUCTION
Glucose, Insulin, and the Glucose Transport System
Glucose is one of the three dietary monosaccharides, along with fructose and galactose, which is directly absorbed into the bloodstream during digestion. This simple monosaccharide serves as both, the main substrate for energy production as well the precursor to the synthesis of many other carbon containing molecules in the body [1].
In relatively healthy individuals, following a meal, glucose absorbed from the gut during digestion stimulates the release of insulin, a peptide hormone, from pancreatic β-cells. This results in glucose uptake by skeletal muscles and adipose tissues, promptly returning plasma glucose levels to the normal range (approximately 4.4-6.1 mM) [1].
During resting conditions, the absorbed glucose is stored as glycogen in the liver and skeletal muscles, and as triglycerides in adipose tissue [1]. In times of fasting or increased energy demand, these stores are quickly re-converted back to glucose and released into the blood stream, helping maintain homeostatic blood glucose concentrations.
By promoting postprandial glucose uptake by muscle and adipose tissues via the Glucose Transport System (GTS) and suppressing hepatic glucose production, insulin is directly responsible for regulating carbohydrate and fat metabolism in the body [1-3].
Given that the cell membrane is impermeable to glucose, specific carrier proteins or transporters spanning the cell membrane allow the binding and uptake of glucose across the hydrophobic lipid bilayer.
Mammalian cells possess two general classes of these carriers/transporters: 1) ATP-dependent Na+/glucose co-transporters that transport glucose against a concentration gradient and 2) facilitative glucose transporters which work in the direction of the glucose gradient [4, 5].
Following digestion, the Na+/glucose co-transporter transports glucose from the lumen of the intestine into polarized epithelial cells where subsequent facilitative transport of glucose moves glucose out of the intestinal epithelium and into the blood. Given that different tissues have different physiological energy needs, their uptake of circulating blood glucose is mediated by tissue-specific glucose transporter proteins called GLUTs [1, 4]. 14 different GLUTs have been identified in mammalian cells that are further divided into three subclasses on the basis of sequence similarities and biochemical properties, of which the roles of GLUT1-GLUT4 in the GTS are the most well characterized [6, 7].
INSULIN-MEDIATED POSTPRANDIAL GLUCOSE TRANSPORT
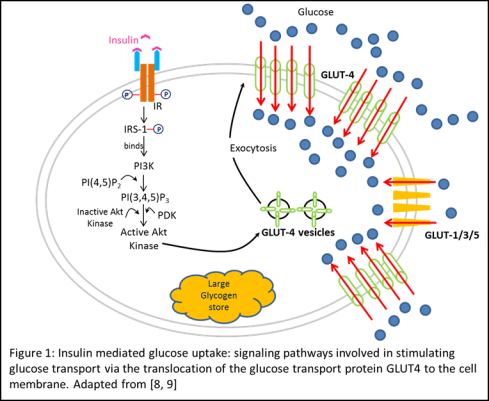
During resting conditions, GLUT1/3/5, constitutively localized to the cell plasma membrane, provide low-level of basal glucose uptake required to sustain respiration in all cells [1]. However, during postprandial conditions, high glucose in the blood stimulates the release of insulin from the β-cells found in the Islets of Langerhans in the pancreas. This secreted insulin regulates the uptake of blood glucose in stimulated skeletal muscles and adipocytes by binding to the insulin receptor (IR) on the surface of the cell (Figure 1). Following insulin binding, the β-subunit of IR undergoes autophosphorylation on tyrosine residues activating its tyrosine kinase activity. The activated IR then phosphorylates Insulin Receptor Substrates (IRS-1) which serves as a docking protein for Phosphatidylinositide 3-kinase (PI3K), activating it. The serine phosphorylation cascade initiated by PI3K activates PI3K-dependent serine/threonine kinases (PDK), which activates downstream Akt kinase, resulting in the translocation and exocytosis of intracellular GLUT4 vesicles to the cell surface. This upregulation of GLUT4 localization on the plasma membrane results in an increased rate of glucose transport into the cell [1, 4, 5, 8, 9].
The necessity of each of these activation steps has been established by in vitro studies on muscle and adipose tissue that used specific kinase inhibitors or mutant proteins and observed the complete ablation of the stimulatory effect of insulin on glucose uptake in insulin stimulated cells. Furthermore, the translocation of GLUT4 specifically in response to insulin – approximately 1% pre- vs. almost 40% post-stimulation, suggests a mechanism of regulation to restrict glucose uptake during low-insulin states [1, 9].
Given that GLUT4 translocation is a critical regulatory site for glucose uptake, abnormal GLUT4 regulation in response to insulin stimulation can have a significant impact on glucose homeostasis in the body. Interestingly, recent studies have shown that mice fed with high caloric diets show significantly decreased glucose uptake in muscle and adipocytes despite stimulation with insulin [8, 10]. Further analysis revealed significant down regulation of PI3K and its downstream substrate PDK in addition to increased expression and activity of Protein Kinase C isoform theta (PKC-θ) in these mice, suggesting impaired insulin mediated signaling and a direct co-relation with high caloric diets (red boxes/arrows in Figure 2) [8, 11-13]. This lack of response to insulin stimulation has long been implicated in the pathogenesis of Type-2 or Non-insulin dependent Diabetes Mellitus (NIDDM) and is commonly referred to as “insulin resistance”.
INSULIN RESISTANCE – FINDING A SWEET SOLUTION
The term “insulin resistance” is used to describe a combination of a lack of insulin-mediated glucose uptake in cells stimulated by insulin and a defect in the ability of insulin to suppress hepatic glucose production; the former commonly attributed to dysregulation of GLUT4 trafficking [14]. Over time, together with reduction in in insulin secretion by the pancreas, these aberrations have been directly linked to causing Type-2 Diabetes mellitus (T2D).
Diminished insulin function at sites of insulin action, such as skeletal muscles and adipocytes (insulin resistance) and a decrease in the ability of pancreatic β-cells to secrete insulin in response to postprandial increase in blood glucose levels, are two hallmarks of T2D [15]. In recent years, Type-2 Diabetes mellitus has emerged as one of the major non-communicable chronic diseases around the world [2]. According to the CDC, that number is currently estimated to be about 26 million in the US alone. Recent epidemiologic data point to excess caloric intake combined with increasingly sedentary lifestyles as factors significantly contributing to this trend [16]. Although both genetic and environmental factors have been implicated in causing these malfunctions, researchers have increasingly suggested that obesity, caused by a chronic imbalance between energy expenditure and energy intake, as one of biggest risk factors for developing insulin resistance and T2D [16].
Given that skeletal muscles constitute approximately 40% of human body mass and have relatively high energy requirements, they account for almost 50-75% of insulin-stimulated glucose uptake in the body [17]. Thus, skeletal muscle is considered the most important tissue with regards to insulin-stimulated blood glucose disposal, and correspondingly maintenance of glucose homeostasis, especially during postprandial periods. Interestingly, both in vitro and in vivo studies dating back to as early as 1987 have shown that in muscle cells, stimuli other than insulin can activate the glucose transport system, namely GLUT4 translocation, to a similar magnitude, albeit likely via a separate (insulin independent) signaling pathway [3, 9, 17].
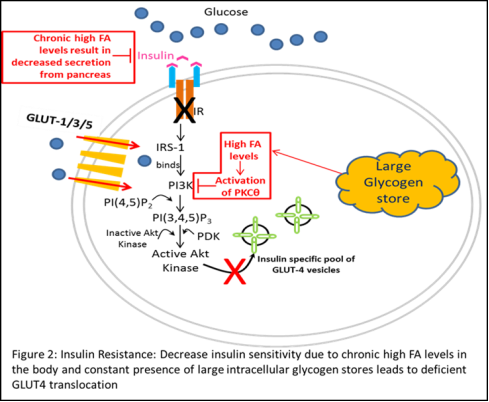
Furthermore, while GLUT4 gene expression was reduced in adipose cells in insulin-resistant states such as obesity and NIDDM, skeletal muscle GLUT4 expression remained normal [9]. This finding prompted the thinking that insulin resistance in skeletal muscle is most likely due to alterations in the translocation, docking, or fusion of glucose transporters at the plasma membrane, and not defects at the transcription or translational levels.
Additionally, researchers observed that in rodents, various modes of exercise and muscle contractile activity such as running exercises, swimming exercises, contraction of hind limb muscles via sciatic nerve stimulation, or contraction of isolated muscles, caused GLUT4 translocation in these skeletal muscles [18]. Additionally, similar studies published by the Goodyear, Holloszy, and Hargreaves groups over the past three years using human models have demonstrated that a single bout of exercise significantly increases glucose transport by upreguatling both GLUT4 expression and translocation in muscles, adding significant support to the hypothesis suggesting that exercise induced muscle contraction can improve glucose uptake by increasing both GLUT4 expression and translocation, potentially reversing the effects of insulin resistance and T2D [3, 5, 10].
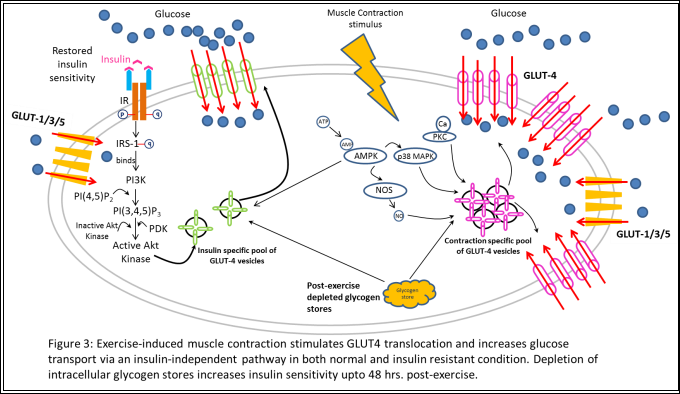
WORKING THE GLUTs – EXERCISE INCREASES GLUT4 TRANSLOCATION AND GLUCOSE TRANSOPORT
Despite the fact that insulin stimulation and exercise-induced muscle contractile activity induce similar magnitudes of increases in muscle glucose transport via upregulation of GLUT4 vesicle translocation to the plasma membrane, the mechanisms of action of these two stimuli are very distinct [19, 20].Studies using cellular fractionation techniques have shown the presence of two distinct intracellular pools of sequestered GLUT4 vesicles, both containing the insulin-responsive amino peptidase (IRAP) and vesicle-associated membrane protein-2 (VAMP-2) that assist in in regulating endocytosis/exocytosis [20, 21]. However, the contraction stimulated GLUT4-vesicle pool was not recruited during insulin stimulation and the important components of the insulin signaling cascade – IR, IRS-1, PI3K, etc., were not phosphorylated or activated by exercise [22-24]. Additionally, Wortmanin, a potent PI3K inhibitor, did not inhibit glucose transport stimulated by muscle contraction as it does the insulin-stimulated pathway, clearly indicating the presence of a distinct pathway [24].
Intracellular calcium, 5’AMP-activated protein kinase (AMPK), nitric oxide (NO), and mitogen activated protein kinase (MAPK) p38, have been implicated regulating GLUT4 translocation in the exercise-medicated effect, although the exact mechanism of how each of these exerts its influence is currently being explored (Figure 3) [9]. These acute effects of exercise, in conjecture with dietary restrictions and pharmaceutical interventions, can and are currently exploited by individuals and pharmaceutical companies in an attempt find a cure to insulin resistance and T2D.
Although the mechanism of how chronic exercise training improves insulin sensitivity is unclear, muscle levels of GLUT4 as well as the activity of glycogen synthase have been shown to be elevated in athletes compared to sedentary controls with muscle GLUT4 expression increasing in individuals who exercise regularly[25]. This likely due to the fact that many genes involved in carbohydrate metabolism (hexokinase, GLUT-4) appear to be jointly upregulated together with mitochondrial enzymes in response to increased muscle contractile activity [10, 18, 19, 22] .
CONCLUSIONS
The recent drastic increase in the prevalence of T2D has been attributed mainly to decreased levels of physical activity and increased caloric intake. Several studies discussed in this paper have shown that higher levels of habitual physical activity results in increased glucose uptake in skeletal muscles via an insulin-independent mechanism. Furthermore, regular exercise has also been shown to improve insulin sensitivity and glucose uptake in subjects with insulin resistance and T2D.
Thus, regular exercise together with diet and weight control, could be the answer to overcoming insulin resistance and significantly reducing the risk of acquiring Type-2 diabetes, and in turn decreasing the odds of developing dilapidating conditions such as atherosclerosis, glaucoma, and stoke.
References
1. Bogan, J.S. (2012). Regulation of glucose transporter translocation in health and diabetes. Annual review of biochemistry 81, 507-532.
2. Oliveira, C., Simoes, M., Carvalho, J., and Ribeiro, J. (2012). Combined exercise for people with type 2 diabetes mellitus: a systematic review. Diabetes research and clinical practice 98, 187-198.
3. Hansen, P.A., Nolte, L.A., Chen, M.M., and Holloszy, J.O. (1998). Increased GLUT-4 translocation mediates enhanced insulin sensitivity of muscle glucose transport after exercise. J Appl Physiol 85, 1218-1222.
4. Stephens, J.M., and Pilch, P.F. (1995). The metabolic regulation and vesicular transport of GLUT4, the major insulin-responsive glucose transporter. Endocrine reviews 16, 529-546.
5. Thorell, A., Hirshman, M.F., Nygren, J., Jorfeldt, L., Wojtaszewski, J.F., Dufresne, S.D., Horton, E.S., Ljungqvist, O., and Goodyear, L.J. (1999). Exercise and insulin cause GLUT-4 translocation in human skeletal muscle. The American journal of physiology 277, E733-741.
6. Joost, H.G., and Thorens, B. (2001). The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review). Molecular membrane biology 18, 247-256.
7. Thorens, B., and Mueckler, M. (2010). Glucose transporters in the 21st Century. American journal of physiology. Endocrinology and metabolism 298, E141-145.
8. Yu, C., Chen, Y., Cline, G.W., Zhang, D., Zong, H., Wang, Y., Bergeron, R., Kim, J.K., Cushman, S.W., Cooney, G.J., et al. (2002). Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. The Journal of biological chemistry 277, 50230-50236.
9. Goodyear, L.J., and Kahn, B.B. (1998). Exercise, glucose transport, and insulin sensitivity. Annual review of medicine 49, 235-261.
10. Hussey, S.E., McGee, S.L., Garnham, A., McConell, G.K., and Hargreaves, M. (2012). Exercise increases skeletal muscle GLUT4 gene expression in patients with type 2 diabetes. Diabetes, obesity & metabolism 14, 768-771.
11. Samuel, V.T., Petersen, K.F., and Shulman, G.I. (2010). Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375, 2267-2277.
12. Malhotra, V., and Campelo, F. (2011). PKD regulates membrane fission to generate TGN to cell surface transport carriers. Cold Spring Harbor perspectives in biology 3.
13. Summers, S.A., Garza, L.A., Zhou, H., and Birnbaum, M.J. (1998). Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Molecular and cellular biology 18, 5457-5464.
14. Tanaka, S., Hayashi, T., Toyoda, T., Hamada, T., Shimizu, Y., Hirata, M., Ebihara, K., Masuzaki, H., Hosoda, K., Fushiki, T., et al. (2007). High-fat diet impairs the effects of a single bout of endurance exercise on glucose transport and insulin sensitivity in rat skeletal muscle. Metabolism: clinical and experimental 56, 1719-1728.
15. Kaufman, R.J. (2011). Beta-cell failure, stress, and type 2 diabetes. The New England journal of medicine 365, 1931-1933.
16. Shuldiner, A.R., Yang, R., and Gong, D.W. (2001). Resistin, obesity and insulin resistance–the emerging role of the adipocyte as an endocrine organ. The New England journal of medicine 345, 1345-1346.
17. Frosig, C., and Richter, E.A. (2009). Improved insulin sensitivity after exercise: focus on insulin signaling. Obesity (Silver Spring) 17 Suppl 3, S15-20.
18. Hayashi, T., Wojtaszewski, J.F., and Goodyear, L.J. (1997). Exercise regulation of glucose transport in skeletal muscle. The American journal of physiology 273, E1039-1051.
19. Douen, A.G., Ramlal, T., Rastogi, S., Bilan, P.J., Cartee, G.D., Vranic, M., Holloszy, J.O., and Klip, A. (1990). Exercise induces recruitment of the “insulin-responsive glucose transporter”. Evidence for distinct intracellular insulin- and exercise-recruitable transporter pools in skeletal muscle. The Journal of biological chemistry 265, 13427-13430.
20. Coderre, L., Kandror, K.V., Vallega, G., and Pilch, P.F. (1995). Identification and characterization of an exercise-sensitive pool of glucose transporters in skeletal muscle. The Journal of biological chemistry 270, 27584-27588.
21. Kristiansen, S., Hargreaves, M., and Richter, E.A. (1996). Exercise-induced increase in glucose transport, GLUT-4, and VAMP-2 in plasma membrane from human muscle. The American journal of physiology 270, E197-201.
22. Treadway, J.L., James, D.E., Burcel, E., and Ruderman, N.B. (1989). Effect of exercise on insulin receptor binding and kinase activity in skeletal muscle. The American journal of physiology 256, E138-144.
23. Goodyear, L.J., Giorgino, F., Balon, T.W., Condorelli, G., and Smith, R.J. (1995). Effects of contractile activity on tyrosine phosphoproteins and PI 3-kinase activity in rat skeletal muscle. The American journal of physiology 268, E987-995.
24. Yeh, J.I., Gulve, E.A., Rameh, L., and Birnbaum, M.J. (1995). The effects of wortmannin on rat skeletal muscle. Dissociation of signaling pathways for insulin- and contraction-activated hexose transport. The Journal of biological chemistry 270, 2107-2111.
25. Youngren, J.F., and Barnard, R.J. (1995). Effects of acute and chronic exercise on skeletal muscle glucose transport in aged rats. J Appl Physiol 78, 1750-1756.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal