Lesch-Nyhan Syndrome: Physiology and Features
| ✅ Paper Type: Free Essay | ✅ Subject: Biology |
| ✅ Wordcount: 2118 words | ✅ Published: 08 Jun 2018 |
Discuss the inheritance, molecular and biochemical defects underlying the clinical features associated with Lesch-Nyhan syndrome.
The Lesch-Nyhan syndrome is a rare heritable disorder of inborn error metabolism of purine which was examined in 1964 by Lesch and Nyhan. They investigated the two brothers with hyperuricemia and neurobehavioral problems and suggested that this disorder involves motor impairment and self-injurious behaviors (Lesch, M and Nyhan, W. L., 1964). The popularity of Lesch-Nyhan syndrome is about 1 in 380000 individuals and this disorder presents solely in male. (Seegmiller, J. E., et al, 1967). It is passed on since the X-linked recessive trait causes genetic mutation followed by the exertion of an enzyme called hypoxanthine guanine phosphoribosyltransferase (HGPRT). In the metabolic condition, Lesch-Nyhan syndrome is recognized by purine overproduction (Nyhan, W. L, et al, 1967) which contributes to increased level of uric acid significantly. The clinical features associate with Lesch-Nyhan syndrome are briefly outlined and the inheritance, molecular and biochemical defects underlying the clinical features are also discussed.
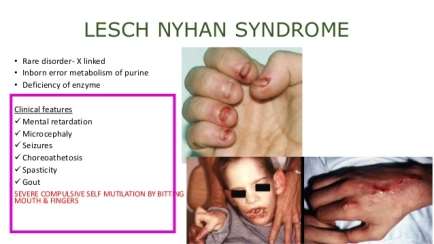
Figure 1. The clinical features associated with Lesch-Nyhan syndrome. This disorder leads to neurobehavioral syndrome involving motor dysfunction, cognitive impairment and self-injurious behaviors (Mumues, 2015).
The clinical characteristics of Lesch-Nyhan syndrome are defined by motor defection leading to neurological operation, cognitive and behavioral impairment, and also overproduction of uric acid or hyperuricemia (van der Zee et al., 1968). Moreover, neurological disturbance is majorly performed as hypotonia and developmental delay which are obviously expressed by three to six months. The children are interrupted in sitting and walking or require a wheelchair in severe condition. Additionally, the twitch of facial muscles and limbs can be observed in elder children. During the first ages, the extrapyramidal difficulties such as dystonia and choreoathetosis or the pyramidal difficulties like spasticity and hyperreflexia become noticeable (Jinnah and Friedmann, 2001). Furthermore, cognitive dysfunction and behavioral impairment occurring between two to three years old can lead to moderate or serious mental hindrance. Also, the continuous self-injurious behaviors such as biting the fingers or lips and hitting the head or limbs are the most distinctive behavioral symptom in patients (Nyhan, W. L, et al, 1968). Lesch-Nyhan syndrome is also characterized by overproduction of uric acid and this induces the decomposition of uric acid crystals in kidneys, ureters or bladder and the gouty arthritis can be developed later in the disorder (Page et al, 1987).
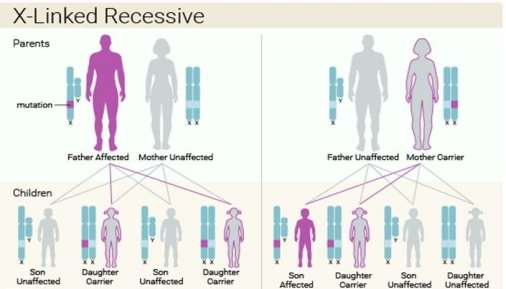
Figure 2. The inheritance detects of Lesch- Nyhan syndrome. This X-linked recessive syndrome is solely for the male and the chance taken from the sons and daughters with either affected father and unaffected mother or unaffected father and carrier mother are showed (IFFA, 2016).
Firstly, the Lesch- Nyhan syndrome is inherited in an X-linked recessive characteristic since the mutation of HPRT gene is positioned on the X chromosome and this disorder is described to the male only (Hoefnagel et al, 1965). More importantly, the X-linked inheritance determined that the X-linked character cannot be transferred from the father to the son. In this syndrome, the Lyon hypothesis can be adopted to demonstrate the mothers are heterozygous and the mosaics involve two cell cultures where one is entirely normal and another one is fully faulty (Migeon et al, 1968) . An investigation is examined that the fibroblasts developing in the cell populations from the skin were duplicated (Migeon, B. R, et al, 1968) and HGPRT deficiency in the negative duplication can be observed through radioautographical method. Furthermore, the halt of the X chromosome is not described as a random activity because HGPRT in the erythrocytes or leukocytes of obliged heterozygotes for this situation is distinct with the glucose 6-phosphate dehydrogenase (G6PD) deficiency where the transportation of enzyme in heterozygotes is around 50%. It is indicated that the enzyme action in the erythrocytes of the mothers with Lesch-Nyhan syndrome are normal. Also, the blood-relative family in two kinds of G6PD and HGPRT (Nyhan, W. L.et al, 1970) are separating so this shows that females were heterozygous to G6PD. The reason why the males have more frequent chance to suffer from this X linked recessive syndrome than the females is because the males contain only one X chromosome and one changed copy of the gene is enough to lead to this syndrome and it is uncommon that females possess two changed copies of the gene Vogel (1977).
It is examined that the father of a male patient is not the carrier of the mutated gene and also do not suffer from the syndrome. The chance taken from the siblings rely upon the carrier condition of the mother. Carrier women contain a 50% chance of passing on the HPRT1 variation in each gestation. The sons who receive that variation will be influenced and the daughters who obtain the variation are served as carriers. Hence, it can be concluded that a carrier mother has a one-fourth chance of getting an affected son, one-fourth chance of getting a carrier daughter and half chance of getting a normal son or daughter (Genetic Home Reference, 2007).
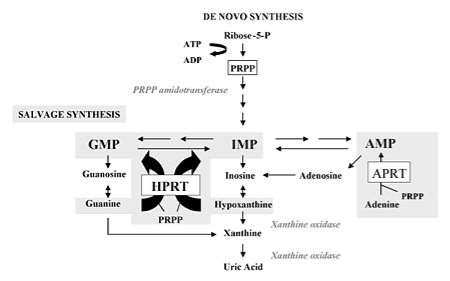
Figure 3. Purine metabolism with de novo synthesis and salvage synthesis. The de novo purine synthesis is mobilized by the enzyme, PRPP amidotransferase, and the salvage pathway by hypoxanthine phosphorybosyltransferase (HPRT) and adenine phosphorybosyltransferase (APRT). HPRT catalyzes the salvage synthesis of inosine monophosphate, IMP, and guanosine monophosphate, GMP, from hypoxanthine and guanine particularly by using PRPP as a co-substrate. The HPRT deficiency leads to decomposition of hypoxanthine and guanine which are transformed into uric acid by xanthine oxidase. Purine overproduction, increase level of PRPP and decrease level of IMP and GMP are the results (Torres R J and Puig J G, 2007).
Secondly, Lesch-Nyhan syndrome contributes to the molecular defects due to the mutation of HPRT1 gene which encrypts hypoxanthine phosphoribosyltransferase. This enzyme is used to recycle purines and is responsible for transferring hypoxanthine and guanine back into DNA synthesis which establish the cells to possess enough supply of building blocks and assemble DNA and RNA (Sweetman, L and Nyhan, W. L. 1972). The deficiency of HPRT map to chromosome Xq26-q27.2 and is likely to cause heterogenous mutations (Greene, M. L, et al, 1970). Also, it induces the increased level of purine nucleotides by purine salvage pathway. It leads to decomposition of uric acid because of the broken down but unrecycled purines and occurrence of excessive PRPP, resulting in the neurological and behavioral problems of Lesch-Nyhan syndrome (Kelley, W. N, 1968).
The de novo pathway is initiated with the active form of ribose from ribose-5-phosphate to 5-phosphoribosyl-1-pyrophosphate (PRPP) and this synthesis discharges AMP. PRPP gives rise to the first nucleotide called inosine monophosphate (IMP) by an enzyme PRPP amidotransferase which serves as the rate-limiting step. Besides, IMP uses as a forerunner of AMP and GMP synthesis and the route of pathway is modulated by the higher level of particular nucleotide (Fox, I. H and Kelley, W. N, 1971). There are two main transferase enzymes involving in the purines salvage pathway. HPRT is responsible for catalyzing the hypoxanthine to inosine monophosphate (IMP) and guanine to guanosine monophosphate (GMP) and another one is adenosine phosphoribosyltransferase (APRT) which assembles adenine to AMP through transferring the 5-phosphoribosyl group from 5-phosphoribosyl 1-pyrophosphate (PRPP) (Keebaugh et al., 2007). The AMP, GMP, IMP are all catabolized to uric acid and particular mononucleotides is started with the transformation to the phosphate free nucleoside via cytosolic 5′-nucleotidases. After that, the nitrogen is eliminated from adenosine to produce inosine with an enzyme, adenosine deaminase (ADA). The ribose is discarded from the nucleotides by purine nucleoside phosphorylase (PNP) and create hypoxanthine, xanthine and guanine (Rubin, C. S.et al, 1969). The nitrogen is then eliminated from guanine through guanine deaminase and xanthine is produced. Eventually, hypoxanthine and xanthine are converted to the final product of purine catabolism called uric acid by xanthine oxidase (Kelley, W. N, et al, 1970). It can be concluded that HGPRT deficiency leads to higher level of PRPP because of its rate limiting function and decreased level of GMP and IMP, resulting in increased de novo purine synthesis and degradation of purines to contribute to the higher level of uric acid called hyperuricemia and cause Lesch-Nyhan syndrome (Rosenbloom, F. M, et al, 1968).
Thirdly, the biochemical defects can be determined by the purine metabolism of the Lesch-Nyhan syndrome and the metabolism can indicate the rate of uric acid synthesizing from 14C-labeled glycine (Sweetman, L. 1968). Also, the deficiency of HPRT contributes to the excessive purine synthesis and this enzyme acts as a regulating role in purine metabolism. The specific activity of the uric acid can be examined by the isolation and purification from urine. In the children with Lesch-Nyhan syndrome it is observed that accelerated specific activity in the uric acid obtain 20 times higher glycine concentration salvaged in uric acid than the control individuals (Nyhan, W. L.et al, 1968). Thus, it leads to the higher level of purine overproduction in patients. Besides, the rate of de novo synthesis of purine using fibroblasts grown in cell culture can be demonstrated (Seegmiller, J. E., et al, 1967). There is a significant relationship between asperity of disorder and recycle of hypoxanthine or guanine. The expressions of intracellular purines are normal within the fibroblasts with deficient HGPRT but purine depletion is apparent since the increased purine metabolites are discharged from cells. In addition, the normal purines were probable to cause a compensative increase in purine synthesis due to notable rising in purinosomes.
Moreover, the purine de novo overproduction showing in this syndrome is an unsuccessful feedback regulation. The first step of the purine pathway is catalyzed by phosphoribosyl pyrophosphate amidotmnsfemse and is dependent to feedback inhibition by adenine and guanine. 2-ethylamino-l,3,4-thiadiazole was used for the examination and the observation showed that uricogenic agent significantly increases the rate of purine synthesis in normal conditions but in the patients the rates of purine synthesis increase more intensely by this agent (Nyhan, W. L.et al, 1968). Furthermore, the existence of xanthinuria in patients with inborn deficiency of xanthine oxidase expresses the abnormalities in the central nervous system. In the cerebrospinal fluid (Sweetman, L. 1968) the xanthine levels are equivalent to the controls and the hypoxanthine level in patients with the Lesch-Nyhan syndrome were greater than the controls by four times. Hence, Hypoxanthine and guanine recycling and the de novo purine synthesis are demonstrated to determine the biochemical abnormality of Lesch-Nyhan syndrome.
In conclusion, by the discussion of the inheritance, molecular and biochemical defects that determining the clinical trait correlated with the Lesch-Nyhan syndrome, it is demonstrated that this X-linked recessive disorder is exclusive for males and is induced from the mutation of HGPRT gene which contributes to the overproduction of uric acid by purine salvage pathway and causes neurobehavioral problems of affected individuals (Rosenbloom, F. M.et al, 1968). For the future perspectives, although there is no treatment to cure this disorder, effective medications can be adopted to alleviate the symptoms such as managing the uric acid levels by allopurinol and reducing behavioral problems by taking diazepam or haloperidol appropriately (Genetics Home Reference, 2007).
(Word count: 1639)
References:
Fox, I. H and Kelley, W. N. 1971. Ann. Intern. Med. 74:424-33 Genetics Home Reference, 2007. Lesch-Nyhan syndrome. Retrieved on 24/5/2007.
Greene, M. L and Boyle, J. A, 1970. Science 167:887- 89
Hoefnagel, D, et al, 1965. Hereditary choreoathetosis, self-mutilation and hyperuricemia in young males. New Eng. J. Med. 273: 130-135, 1965.
IFFA, 2016. Lesch-Nyhan Syndrome on emaze. [digital image] Retrieved from: https://www.emaze.com/@ACFTIZWL/Lesch-Nyhan-Syndrome
Jinnah, H. A and Friedmann, T. 2001. Lesch-Nyhan disease and its variants.In: Scriver, C. R.; Beaudet, A. L.; Sly, W. S.; Valle, D. (eds.): The Metabolic & Molecular Bases of Inherited Disease. Vol. II. (8th ed.) New York: McGraw-Hill (pub.) P. 2537.
Kelley, W. N, 1968. Fed. Proc. 27: 1047-52
Keebaugh, A. C and Sullivan, R. T, 2007. NISC Comparative Sequencing Program, Thomas, J. W. Gene duplication and inactivation in the HPRT gene family. Genomics 89: 134-142.
Kelley, W. N, et al, 1970. Biochim. Biophys. Acta 21 5 :512-16
Lesch, M and Nyhan, W. L, 1964. Am. J. Med. 36:561-70
Mumues, 2015. Mental retardation. [digital image] Retrieved from:
https://www.slideshare.net/mumues/mental-retardation-44326087
Migeon, B. R, et al, 1968. X-linked hypoxanthine-guanine phosphoribosyl transferase deficiency: heterozygote has two clonal populations. Science 160: 425-427.
Nyhan, W. L, 1967. Pediat. 67 :257-63
Nyhan, W. L, et al, 1968. Metabolism 17 :846-53
Nyhan, W. L, et al, 1970. Proc. Nat. Acad. Sci. USA 65:214-18
Page, T, et al, 1987. Syndrome of mild mental retardation, spastic gait, and skeletal
malformations in a family with partial deficiency of hypoxanthine-guanine
phosphoribosyltransferase. Pediatrics 79: 713-717
Rubin, C. S, et al, 1969. J. Lab. Cfin. Med. 74:732-41
Rosenbloom, F. M, et al, 1968. Bioi. Chern. 243 : 1 166-73
Seegmiller, J. E, et al, 1967. Science 155: 1 682-84
Sweetman, L. 1968. Fed. Proc. 27 : 1 055-59
Sweetman, L and Nyhan, W. L. 1972. Arch. Intern. Med. 130:214-20
Torres R J and Puig J G, 2007. Hypoxanthaine-guanine phosphoribosyltransferase
(HPRT) deficiency: Lesch- Nyhan Syndrome. Orphanet J Rare Dis. 2, 1. [digital image]
Retrieved from: https://en.wikipedia.org/wiki/Lesch%E2%80%93Nyhan_syndrome#/media/File:HPRT_metabolism.jpg
Vogel, F, 1977. A probable sex difference in some mutation rates. (Editorial) Am. J.
Hum. Genet. 29: 312-319.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal