Tissue and Cellular Stiffening in Maladaptive Vascular Smooth Muscle Cell Phenotypic Change
| ✅ Paper Type: Free Essay | ✅ Subject: Sciences |
| ✅ Wordcount: 1853 words | ✅ Published: 23 Sep 2019 |
The role of tissue and cellular stiffening in maladaptive vascular smooth muscle cell phenotypic change.
- Cardiovascular Disease
Cardiovascular disease (CVD) refers to a heterogeneous group of diseases afflicting the heart and vasculature with similar underlying pathophysiological manifestations, such as coronary heart diseases, cerebrovascular disease, hypertension, peripheral arterial disease, congenital heart diseases and heart failure. The prevalence of CVD had reached global epidemic proportions, with an estimated 422.7 million cases worldwide [Figure 1]1. CVD is the universal leading cause of mortality, resulting in 17.9 million deaths in 2015, accounting for 44% of all non-communicable disease deaths2. In the UK, around 7 million people are living with CVD and 25% of all deaths (>150,000 deaths/annum) are attributable to CVD3.
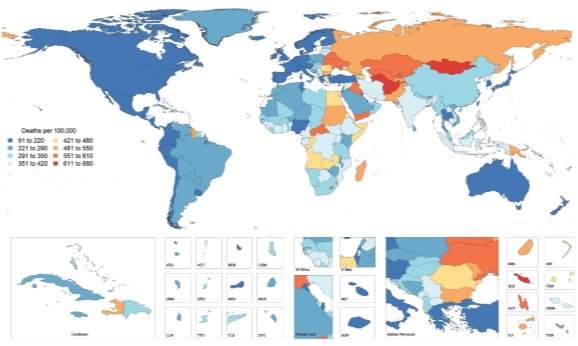
Figure 1: Global Map of Age-Standardised Death Rate of CVD in 2015. Choropleth map illustrating the estimated age-standardized mortality rate of total CVD in 2015 for each country1.
The worldwide burden of CVD on public health is escalating due to global demographic and socio-economic trends, particularly population ageing, economic growth in Asia and the obesity epidemic observed in developed countries. In addition, more than 75% of CVD related deaths occur in low- and middle-income countries, as a consequence of restricted access to primary healthcare and affordable treatments. Thus, an enhanced understanding of cardiovascular physiology and CVD mechanisms, as well as the development of novel, more targeted and low-cost prevention, early diagnostic and therapeutic strategies are required to reduce the incidence of CVD mortality4.
CVD pathogenesis and related complications are attributed to numerous risk factors including metabolic factors like hypertension, hyperglycaemia, dyslipidaemia and obesity, coupled with behavioural factors such as imbalanced diet, physical inactivity, excessive alcohol consumption and tobacco use. Ageing is an integral, independent risk factor for CVD5, however many questions are unanswered concerning ageing and CVD. Here, the focus will be on elucidating possible links between ageing, CVD and vascular calcification.
- Ageing
- Arterial Stiffening
Arterial compliance is dependent upon intrinsic material stiffness and the arterial architecture. Arterial stiffness can be defined as a diminishing distensibility that characterises the relative changes occurring in the lumen cross-sectional area for a given variation in blood pressure18.
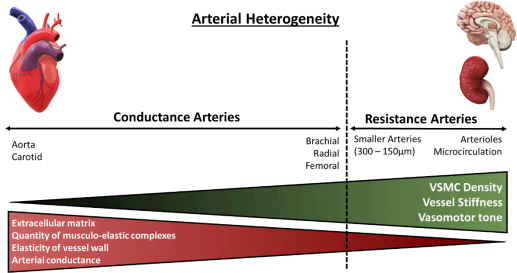
The distensibility or stiffness of the arteries contributes to wave propagation and reflection in the arterial vasculature, where the arterial pulse disseminates with a certain speed known as the pulse wave velocity (PWV), continuously varying in amplification and shape. Arterial stiffness has aetiological implications in various cardiovascular diseases and has been identified as a key independent risk factor for all-cause and CVD mortality19,20. Aortic PWV is a reference parameter largely accepted as the simplest, non-invasive, accurate and reproducible measurement of central arterial stiffness for large elastic arteries. Clinically, the carotid-femoral PWV is considered as the ‘gold-standard’ measurement to determine arterial stiffness, measured along the aorto-iliac pathway and has been previously utilised to determine the predictive value of aortic stiffness and cardiovascular incidence21. Arterial stiffness indexes, for example, Young’s elastic modulus calculated from stress-strain curves, are utilised for smaller-sized muscular arteries. Both central and peripheral stiffness is influenced by the intricate interactions of the intramural cells and ECM, which are responsible for the mechanical function and structural integrity of the vasculature, differing with vessel size22. The aetiology of arterial stiffening is attributed to various molecular and cellular determinants including key ECM components (primarily elastin and collagen fibres), cell-ECM interactions and VSMC tone regulatory proteins. In addition, mechanical factors of arterial wall remodelling (shear stress and pulsatile, circumferential stress) and VSMC plasticity are key determinants of arterial stiffness18.
Figure 2: Architectural and functional divergence of the arterial vasculature. The diversity of the arterial vasculature ensures that conductance and resistance arteries can exert their respective functions. Larger elastic arteries, known as conductance arteries, convert pulsatile flow and pressure into continuous pressure, enabling oxygen delivery with nominal energy loss in arteriolar vascular walls. Resistance arteries are smaller in diameter, include arterioles and the microcirculation, and are responsible for the distribution of blood flow to target organs. Red triangle: Demonstrates arterial compliance of various arteries, amount of ECM present within vessels’ wall and number of musculo-elastic complexes. Green triangle: Demonstrates density of VSMCs within arterial wall, vessel stiffness and vasomotor tone, the function of which augments with diminishing vessel size. Adapted from Lacolley, P. et al (2017)14.
- Smooth muscle cells
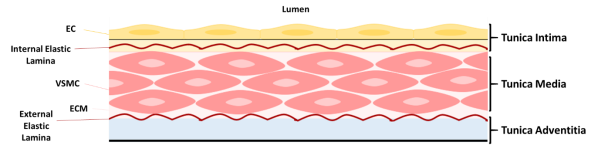
Smooth muscle is a non-striated, involuntary muscle that is distributed mainly in the hollow and tubular organs, for example, bladder, trachea, digestive tract and the vasculature. Smooth muscle consists of mononucleated, elongated and fusiform cells, which produce copious amounts of extracellular matrix (ECM)6. The principal function of smooth muscle cells (SMCs) is to contract and produce mechanical output required for specific organ function. Vascular smooth muscle cells (VSMCs) are stromal, contractile cells and the predominant cell type found in the tunica media of blood vessels [Fig.2]7. In addition, VSMCs maintain and remodel the ECM in the vasculature8.
Figure 3: Structure of blood vessel wall. The tunica intima consists of endothelial cells (ECs) resting on the internal elastic lamina. The tunica media comprises of elastin and collagen fibres and loosely dispersed, circumferentially aligned vascular smooth muscle cells (VSMCs) in extracellular matrix (ECM), embedded between the internal and external elastic laminae. VSMCs are attached to the elastic laminae by dense plaques, converging to focal adhesion complexes. Collagen fibres occur laterally to the elastic laminae. Oxytalan fibres, composed of fibrillin, connect VSMCs to the elastic laminae. The tunica adventitia is composed of collagen fibres and fibroblasts. Adapted from Lacolley, P. et al (2017)and Ponticos, M and Smith, B (2014).
VSMCs demonstrate a diverse mosaic developmental pattern, deriving from various embryogenic tissues such as the proepicardium, mesothelium, neural crest and somites9. Flk1-positive embryonic stem cells serve as vascular progenitors, for endothelial cells and perivascular mural cells, including VSMCs and pericytes10. The primary function of VSMCs is to provide structural stability, functionality and to resist mechanical stress in the vasculature effectuated by the high-pressure load of circulating blood. Through coordinated contraction and relaxation cycles in opposition to the heart, VSMCs adjust vessel diameter, regulate haemodynamics and prevent physical permeability in large vessels, ensuring mechanical compliance and sustaining normal blood pressure11,12.
4.1. Smooth muscle contraction and relaxation
SMC contraction/relaxation signalling and mechanics are coordinated by the 20kDa myosin light chain protein (MLC20). Active MLC20 stimulates myosin Mg2+ ATPase function, enabling its binding and sliding along the actin filament, concluding in cell and muscle contraction. There are two main contraction signalling mechanisms in SMCs. The Ca2+-calmodulin system, which requires Ca2+-dependent activation of MLC20 and is mediated by myosin light chain kinase (MLCK). An influx of extracellular Ca2+ through plasmalemmal Ca2+ channels and the release of sarcoplasmic reticulum Ca2+ precipitates in an incline of free cytoplasmic Ca2+, resulting in the activation of the Ca2+-calmodulin system. Calmodulin and intracellular Ca2+ form complexes, activating MLCK and initiating SMC contraction. Vasoactive ligands include vasopressin, angiotensin II, endothelin-1, norepinephrine and prostaglandins.
Ca2+-sensitisation is the second contraction signalling mechanism. It is Ca2+ independent, facilitated by the RhoA/Rho-associated protein kinase (ROCK) pathway and its primary function is the maintenance of force generation in the absence of adequate intracellular Ca2+ concentrations. RhoA recruits and activates ROCK, which in turn, phosphorylates MLC phosphatase and MLC20, stimulating SMC contraction. Relaxation is accomplished through the elimination of contractile stimuli, for example, the decline of free cytoplasmic Ca2+ and/or the direct action of vasodilator agonists, including natriuretic peptides, nitric oxide, insulin and adenosine13–16.
4.2. VSMC Plasticity
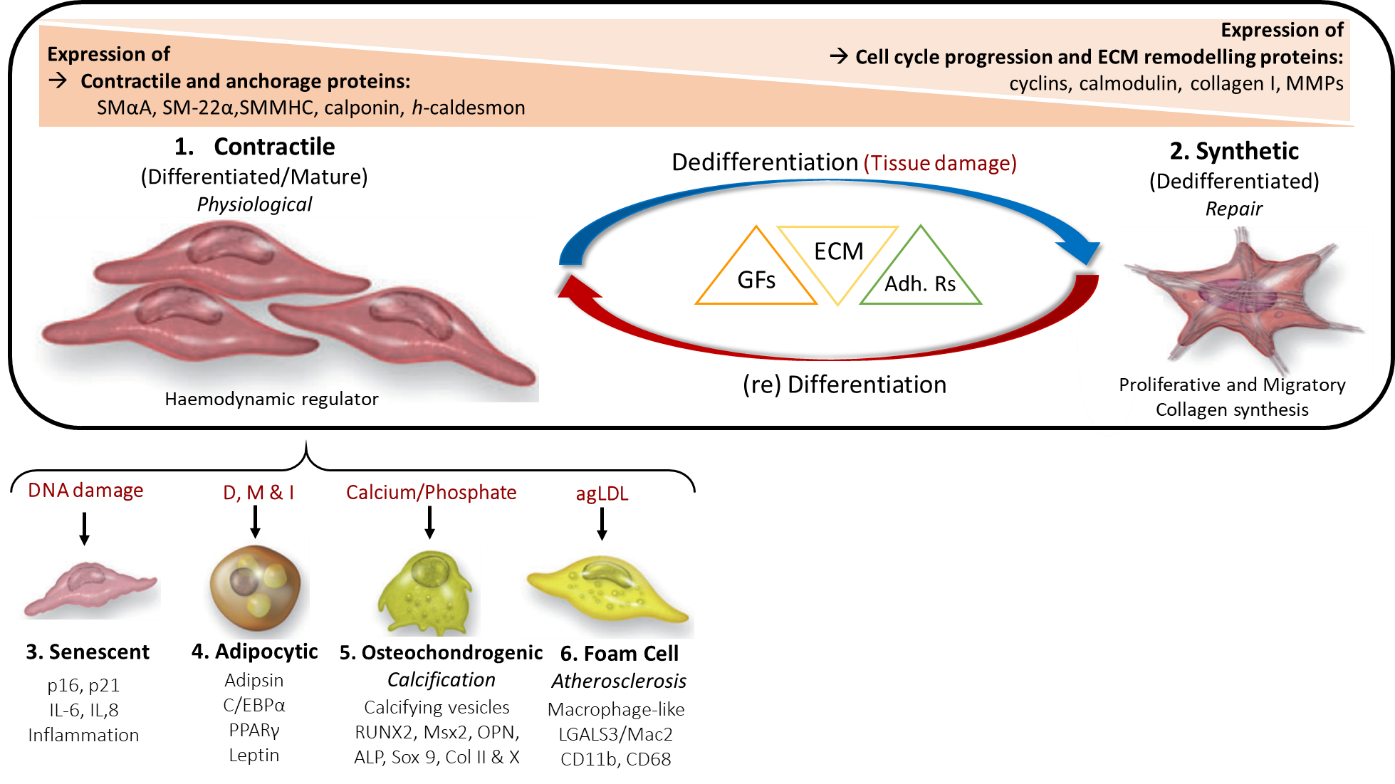
Under normal physiological conditions, mature VSMCs are phenotypically contractile and differentiated, with a slow proliferation rate. They respond to stimulatory neurotransmitters (e.g. acetylcholine) and express various contractile and anchorage markers including smooth muscle α-actin (SMαA), SM-22α, SM myosin heavy chains SM-1, SM-2 (SMMHC-1,-2) calponin, h-caldesmon and smoothelin17. However, VSMCs are not terminally differentiated and display functional phenotypic plasticity, which is exhibited in response to vascular injury or local environmental signals (e.g. paracrine signals, cell-cell and/or cell-matrix interactions). As a result of tissue damage, VSMCs modulate their phenotype and acquire functional properties such as migratory, synthetic and proliferative, which in turn, facilitate tissue repair.
Repair and renewal : Mature VSMCs contribute to smooth muscle and vessel repair due to their enhanced cellular plasticity, facilitating phenotypic switching from differentiated (contractile-quiescent) to dedifferentiated (synthetic and proliferative-migratory).
- Vascular Calcification
VSMCs are the fundamental cell type implicated in vascular calcification.
Points to write about:
- Talk about morphological changes observed in aorta of young and aged rats (Abu-Dief (2016))
References ADD URL and Date for WHO and BHF references
- Roth, G. A. et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 70, 1–25 (2017).
- WHO. Cardiovascular Disease Statistics. (2017).
- BHF. British Heart Foundation UK Factsheet (BHF estimate based on latest Quality & Outcomes Framework prevalence data). (2018).
- World Health Organization. World health statistics. 2016, Monitoring health for the SDGs, Sustainable Development Goals.
- North, B. J. & Sinclair, D. A. The intersection between aging and cardiovascular disease. Circ. Res. 110, 1097–108 (2012).
- Kao, C., & Carsten, M. (Eds. . Cellular Aspects of Smooth Muscle Function. (Cambridge University Press, 1997). doi:10.1017/CBO9780511759383
- Metz, R. P., Patterson, J. L. & Wilson, E. Vascular smooth muscle cells: Isolation, culture, and characterization. Methods Mol. Biol. (2012). doi:10.1007/978-1-61779-523-7_16
- Ponticos, M. & Smith, B. D. Extracellular matrix synthesis in vascular disease: hypertension, and atherosclerosis. J. Biomed. Res. 28, 25–39 (2014).
- Majesky, M. W., Dong, X. R., Regan, J. N. & Hoglund, V. J. Vascular smooth muscle progenitor cells: building and repairing blood vessels. Circ. Res. 108, 365–77 (2011).
- Yamashita, J. et al. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 408, 92–96 (2000).
- Frismantiene, A., Philippova, M., Erne, P. & Resink, T. J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell. Signal. 52, 48–64 (2018).
- Dart, A. M. & Kingwell, B. A. Pulse Pressure-A Review of Mechanisms and Clinical Relevance. J. Am. Coll. Cardiol. 37, 975–984 (2001).
- Touyz, R. M. et al. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 114, 529–539 (2018).
- Eto, M. & Kitazawa, T. Diversity and plasticity in signaling pathways that regulate smooth muscle responsiveness: Paradigms and paradoxes for the myosin phosphatase, the master regulator of smooth muscle contraction. doi:10.1540/jsmr.53.1
- Brozovich, F. V et al. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 68, 476–532 (2016).
- Berridge, M. J. Smooth muscle cell calcium activation mechanisms. Journal of Physiology (2008). doi:10.1113/jphysiol.2008.160440
- Durham, A. L., Speer, M. Y., Scatena, M., Giachelli, C. M. & Shanahan, C. M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovascular Research (2018). doi:10.1093/cvr/cvy010
- Lacolley, P., Regnault, V., Segers, P. & Laurent, S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol. Rev. 97, 1555–1617 (2017).
- 19. Ben-Shlomo, Y. et al. Cardiometabolic Risk Aortic Pulse Wave Velocity Improves Cardiovascular Event Prediction An Individual Participant Meta-Analysis of Prospective Observational Data From 17,635 Subjects. J. Am. Coll. Cardiol. 63, 636–646 (2014).
- 20. Laurent, S. et al. Aortic Stiffness Is an Independent Predictor of All-Cause and Cardiovascular Mortality in Hypertensive Patients. Hypertension 37, 1236–1241 (2001).
- 21. Laurent, S. et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications on behalf of the European Network for Non-invasive Investigation of Large Arteries. Eur. Heart J. 27, 2588–2605 (2006).
- 22. Humphrey, J. D., Dufresne, E. R. & Schwartz, M. A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 15, 802–812 (2014).
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal