Analytical Test Methods in Downstream Processing
| ✅ Paper Type: Free Essay | ✅ Subject: Sciences |
| ✅ Wordcount: 3511 words | ✅ Published: 15 Sep 2017 |
Andrea Waldvogel
Validation of Analytical Test Methods in Downstream Processing
Introduction
Quality, safety and efficacy are the main principles of quality assurance of biopharmaceutical drug products. Quality must be designed into the product or process since it cannot be tested into it. Therefore, a quality system must comprise of validation, change control, training, quality control and vendor assurance amongst others.1-3
An effective validation does not only provide a high degree of confidence that the finished drug product consistently and reliably meets all quality requirements but also leads to economic benefits by reducing the cost associated with process monitoring, sampling and testing.4
Biopharmaceutical companies must perform facility, utility and equipment validation/qualification, process validation, computer and computer systems validation, cleaning validation and analytical method validation.5 This project report will focus on analytical method validation, also referred to as analytical procedure validation.
Analytical methods are developed to measure characteristics such as molecular identity, purity, potency, and safety of raw materials, in-process samples and final drug products and to monitor the manufacturing process. The number of tests should be adequate to show manufacturing consistency and the impact of changes on the quality of the drug product. All methods must be demonstrated to be fit for their intended purpose before they are employed.5,6
Analytical method validation means “establishing documented evidence that provides high degree of assurance that a specific method, and the ancillary instruments included in the method, will consistently yield results that accurately reflect the quality characteristics of the product tested”.7
This report begins by providing an overview over some of the regulations and guidelines related to analytical method validation. The second section introduces the modern lifecycle approach to method validation and section three gives an insight into analytical method validation in biopharmaceutical downstream processing. The final section concludes the report with a summary of the main points discussed.
There are many different regulations, guidelines and pharmacopeial monographs concerned with analytical method validation. As it would go beyond the scope of this document to write about all of them, the report focuses on some to give an overview.
1.1 Regulations
Validation is based on, but not prescribed by regulatory requirement. It is best viewed as an essential and integral part of Good Manufacturing Practice (GMP) for the assurance of quality. Compliance with validation requirements is necessary for obtaining approval for clinical trials and to market new products.4
In the U.S. for example, 21 CFR Part 211.165(e) states8:
The accuracy, sensitivity, specificity, and reproducibility of test methods employed by the firm shall be established and documented. Such validation and documentation may be accomplished in accordance with § 211.194(a)(2).
21 CFR Part 211.194(a) (2)8:
A statement of each method used in the testing of the sample. The statement shall indicate the location of data that establish that the methods used in the testing of the sample meet proper standards of accuracy and reliability as applied to the product tested. (…). The suitability of all testing methods used shall be verified under actual conditions of use.
The requirement of validation is also implied in 211.100(a)8:
There shall be written procedures for production and process control designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess.
1.2 Guidelines
The first guidance documents on analytical method validation were published in the 1990s. In the course of time, a lot of revision activity has taken place allowing the incorporation of new approaches to science.
The harmonised ICH Q2(R1) Validation of Analytical Procedures: Text and Methodology guideline, issued in 2005, is considered the primary reference for recommendations and definitions on validation characteristics for analytical procedures and has tended to take on the role of a regulatory expectation. In the United States, it has been used as a guidance along with the related compendial documents USP <1225> Analytical Procedure Validation, <1226> Analytical Procedure Verification, and <1224> Analytical Procedure Transfer. However, those documents do not provide support for the users to accurately understand and control sources of variability.6,9
In 2013, a Stimuli to the Revision Process paper on Lifecycle Management of Analytical Procedures published by the USP Validation and Verification Expert Panel proposed a Quality by Design (QbD) approach to method development, validation, and performance verification of an analytical method via a lifecycle concept. They suggested that the traditional approaches outlined in the U.S. Pharmacopeial monographs <1224>, <1225>, and <1226> should be revised and assembled into a single new general information chapter <1220> Lifecycle Management of Analytical Procedures and a new general chapter <220> specifying the basic requirements. This would, for the first time, formally link method development and method validation within pharmacopeia.6,10
In 2016, a general chapter prospectus on <1220> The Analytical Procedure Lifecycle was posted on the U.S. Pharmacopeial Notices and a draft of a new USP General Chapter <1210> Statistical Tools for Procedure Validation was published in the U.S. Pharmacopeial Forum (U.S. Pharmacopeial Convention).11,12
In August 2017, a new general USP Chapter <1225> Validation of Compendial Methods will become official. This is an effort to better align the validation concept with the revised FDA guidance for industry Analytical Procedures and Methods Validation for Drugs and Biologics issued in 2015. However, instead of including a section on Lifecycle Management of Analytical Procedures only a reference has been added. Depending on the development of the chapters <1220> and <1210>, USP <1225> may be revised again.13
Growing awareness that the implementation of an analytical method with adequate quality steps designed into the procedure during the development phase led to the development of a lifecycle approach for analytical procedure validation.10
2.1 Stages of the Modern Lifecycle Approach
The modern lifecycle approach is based on the Quality by Design (QbD) approach outlined in ICH Q8(R2) guideline and defines activities and deliverables for every stage of method validation. The following diagram provides an overview.
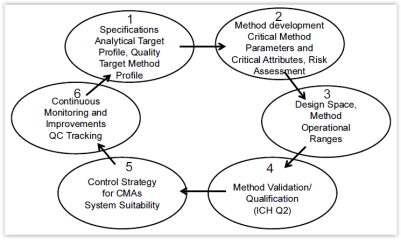
Figure 1: QbD Approach for Analytical Methods
2.1.1 Stage 1: Procedure Design, Development, and Understanding
To be able to design quality into a method to ensure that the method is reliable and meets the analytical target profile (ATP) defined at the beginning of this stage, an understanding of how the procedure works is key. Risk assessment should be undertaken to identify variables that could have an influence on the method. The knowledge of variables and their impact is not only important for the development of a control strategy but also for the determination of a design space. The design space will reduce the amount of revalidation work considerably when the method is used operationally. Key elements of this stage are shown in Figure 1 no. 1-3.10
Proper method development including the evaluation of robustness is essential for an effective analytical procedure.10 Robustness is a measure of the “method’s capacity to remain unaffected by small variations in method parameters and provides an indication of its reliability during normal usage”.9 At this stage, system suitability parameters are established which help to ensure that the analytical method remains valid whenever used.9
Without developing a robust method and an understanding of how a change of key parameters will impact its performance, the actual method validation step will be difficult.10
2.1.2 Stage 2: Procedure Performance Qualification
The lifecycle approach uses the term procedure performance qualification instead of method validation.
Procedure performance qualification is “the verification of the performance of the analytical procedure (either a new one or a revised procedure) against the requirements of the ATP”.10
If the procedure development has been done correctly, this step should simply be a confirmation that it is fit for the intended purpose. In cases where further controls need to be added to ensure reliable results the analytical control strategy, developed during stage 1, will need an update.10
Method validation work should be performed by a user laboratory under the same conditions as it will be used to comply with existing GMP regulations.10
2.1.3 Stage 3: Implementation and Continued Procedure Performance Verification
This stage involves checking how the procedure works during operational use and that it remains in a state of control.10
For this purpose, inputs on reliability and performance of the method gathered from operators and customer complaints will be evaluated. Performance indicators such as system suitability, quality control samples and out-of-specification (OOS) results are tracked and trended.10
The method should be continually improved through corrective and preventive action to reduce the number of out-of-specification (OOS) results. Any change to improve the overall performance needs to be assessed using change control procedures. As shown in Figure 3, the nature of the change specifies what actions have to be taken.6,10,14

Figure 2: Change Types and appropriate Actions
2.2 Traditional (Current) Approach vs Lifecycle Approach
In their Stimuli to Revision paper, the USP Expert Panel recommends the adoption of a lifecycle approach for the management of analytical procedures. In their conclusion, they outline the advantages of a lifecycle approach by comparing it to the traditional (current) approach to analytical procedure validation (Figure 1).6
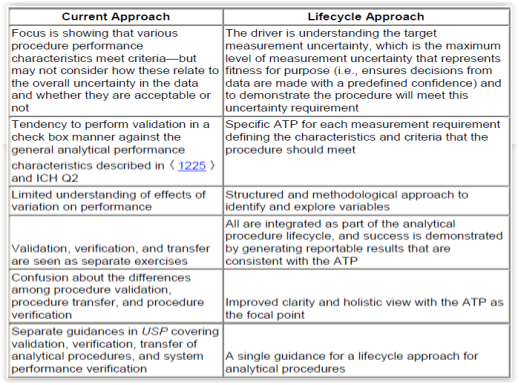

Downstream processing in biopharmaceutical manufacturing involves many steps from recovery over purification to fill finish. Next to in-process monitoring of process parameters such as pH and temperature, analytical testing for the determination of quantity, identity, strength, potency, purity (product- and process related impurities), bioburden and endotoxin has to be performed on raw materials, intermediates, drug substances and finished drug products. Some of the analytical methods in downstream processing are HPLC, gel electrophoresis, PCR, ELISA, Bradford, hemagglutination (HA) and plaque assay.
All critical steps in a process have to be validated and less critical steps have to be under control. The criticality of an analytical method is determined by risk assessment. There are various ways to perform method validation. The manufacturer is responsible for choosing the suitable validation procedure and justifying it.1,7,9
3.1 Types of Analytical Procedures
The four most common types of analytical procedures are identification tests, qualitative and quantitative tests for impurities and assay. Assay involves the quantitative measurement of the major component(s) in the drug substance and drug product.9
3.2 Team Selection
The validation project manager is responsible for the selection of a Cross-Functional-Team (CFT) from various related departments and functional areas. He or she is also in charge of assigning responsibilities and assuring that all personnel involved are trained properly.7
3.3 Analytical Method Validation Protocol
The first step in method validation is the preparation of a protocol that defines the work to be done to demonstrate that the method is fit for its intended use.7,10
 The analytical method validation protocol should contain the following sections:
The analytical method validation protocol should contain the following sections:
|
Purpose |
|
|
Overview |
|
|
Resources |
|
|
Appendices |
|
Before the method validation can begin the protocol must be agreed upon by the CFT and approved.7
3.4 Performance Characteristics Tests
Performance characteristics and their acceptance criteria are defined during the characterisation studies at the development stage of the analytical method. Depending on the method and its intended use, some performance characteristics tests may be omitted, the number of replicates may be increased or reduced, or acceptance criteria may be adapted. All decisions have to be based on scientifically sound judgment. It is important that well characterised reference materials, with documented purity, are used for testing performance characteristics.7,9
The following table outlines the performance characteristics and their meaning, test procedures, how data should be reported and acceptance criteria according to ICH Q2(R1) and FDA Guidance for Industry on Analytical Procedures and Method Validation.
|
Accuracy |
Closeness of test results to the true value |
For drug substances, accuracy measurements are obtained by comparing test results to the analysis of a standard reference material or to a second, well-characterized method. For drug products, accuracy is evaluated by analysing synthetic mixtures (containing all excipient materials in the correct proportions) spiked with known quantities of analyte. Guidelines recommend that data be collected from a minimum of nine determinations over at least three concentration levels covering the specified range. The data should be reported as the percent recovery of the known, added amount, or as the difference between the mean and true value with confidence intervals (such as ±1 SD). Acceptability criteria are defined by end users but rarely fall outside 97-103% of the nominal value. Statistical analysis can be applied using a one sample t-test. |
|
Precision |
Degree of agreement among test results when the method is applied repeatedly to multiple samplings of a homogeneous sample |
Precision is commonly described in terms of repeatability, intermediate precision, and reproducibility:
|
|
Specifity |
Ability to measure accurately and specifically the analyte of interest in the presence of other components |
In drug assays, specificity takes into account the degree of interference from other active ingredients, excipients, impurities, degradation products, or matrices. In chromatography, it ensures that a chromatographic peak corresponds to a single component. Specificity can be demonstrated by the resolution between peaks of interest. |
|
Limit of detection (LOD) |
Lowest concentration of an analyte in a sample that can be detected |
In a chromatography laboratory, the most common way to determine both the LOD and the LOQ is using signal-to-noise ratios (S/N), commonly 3:1 for LOD and 10:1 for LOQ. An appropriate number of samples must be analyzed to fully validate the method performance at the limit. |
|
Limit of quantitation (LOQ) |
Lowest concentration of an analyte in a sample that can be quantified with acceptable precision and accuracy under the stated operational conditions of the method |
|
|
Linearity |
Ability of a method to provide results that are directly proportional to analyte concentration within a given range |
Guidelines specify that a minimum of five concentration levels be used to determine the range and linearity, along with certain minimum specified ranges depending on the type of method. The range is normally expressed in the same units as the test results obtained by the method (for example, nanograms per millilitre). Data to be reported generally include the equation for the calibration curve line, the coefficient of determination (r 2), residuals, and the curve itself. |
|
Range |
Interval between the upper and lower concentrations of an analyte that have been demonstrated to be determined with acceptable precision, accuracy, and linearity using the method |
|
|
Robustness |
Measure of a method’s capacity to obtain comparable and acceptable results when perturbed by small but deliberate variations in procedural parameters |
It provides an indication of the method’s suitability and reliability during normal use. During a robustness study, method parameters (such as eluent composition, gradient, and detector settings) are intentionally varied to study the effects on analytical results. Common chromatography parameters used to measure and document robustness include critical peak pair resolution (R s), plate number (N) or peak width in gradient elution, retention time (t R), tailing factor (T F), peak area (and height) and concentration. Robustness studies are expected to be done during method development. |

Table 7 gives an overview of the performance characteristic tests that have to be performed on different types of analytical procedures.9
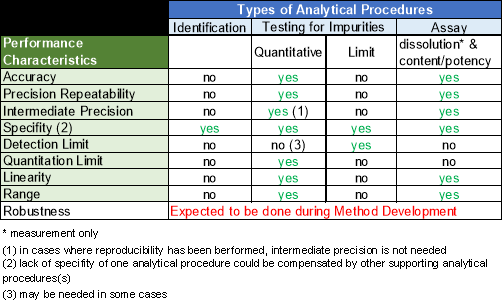
Figure 6: Performance Characteristic tests performed on different Types of Analytical Procedures
The performance characteristics are evaluated by comparing the results to the specifications defined at the development stage.
An analytical method is considered to be validated when it meets the specifications defined at the development stage. Once an analytical method has been made a formal part of the manufacturing process, it is extremely difficult to remove it. In the event of changes in the drug substance, the composition of the finished product and in the analytical procedure, revalidation may be necessary.5,7,9
3.5 Validation Documentation
Every validation step needs to be documented to be able to provide written evidence to the regulatory authorities that a specific method is fit for its purpose. Documentation associated with method validation are validation protocols, standard operating procedures (SOPs), specifications and validation reports.
Downstream processing in biopharmaceutical manufacturing involves many analytical methods which help to ensure quality, safety and efficacy of the final drug product. Development, validation and control of a robust analytical method is a lengthy and difficult task. However, without written evidence that an analytical method is fit for its intended use the company will not obtain a marketing authorisation.
Over time, many guidelines and pharmacopeial monographs have been issued and a lot of revision activity has happened especially following the Stimuli to Revision paper published in 2013. Even though, no comprehensive guideline or monograph incorporating the modern lifecycle approach has been issued yet.
Although proper development of robust and effective analytical methods is more time-consuming and expensive, it has many advantages. It leads to more efficient validation, variability is reduced and controlled and analytical method-related out-of-specification results and failure investigation are minimised. Additionally, changing method parameters within the design space facilitates continual improvement as it does not require regulatory re-approval.
Validation is a team effort. Members of the CFT need to be properly trained. Their first and most demanding task is the preparation of a protocol which defines the scope of the validation project and provides all details necessary for a successful validation. It also defines, depending on the type of the analytical procedure, which performance characteristics need to be tested. The use of well characterised reference materials with known purity is important. Analytical method validation is considered to be complete when all acceptance criteria are met and a validation report has been written.
1 Choudhary, A. (2009). Validation in Pharmaceutical Manufacturing. Pharmaceutical Guideline. [Accessed on 1 March 2017]. Available on Internet: http://www.pharmaguideline.com/2010/12/validation.html
2 International Conference on Harmonization (2009). Harmonised Tripartite Guideline: ICH Q8(R2) Pharmaceutical Development. [Accessed on 1 March 2017]. Available on Internet: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf
3 Stockbridge, P. (2008). Biopharmaceutical Fill and Finish: Technical and Operating Challenges for the Latest Formulations and Devices. BioProcess International. [Accessed on 7 March 2017]. Available on Internet: http://www.bioprocessintl.com/2008/biopharmaceutical-quality-assurance-184041/
4 Nandhakumar, L.; Dharmamoorthy, G.; Rameshkumar, S.; Chandrasekaran, S. (2011). An Overview of Pharmaceutical Validation: Quality Assurance View Point. IJRPC, 1(4). [Accessed on 1 March 2017]. Available on Internet: http://www.caidat.org/m4atomp3/2561456335400862.pdf
5 Lutz, H. (2005). Introduction to Validation of Biopharmaceuticals. BioPharm International. [Accessed on 1 March 2017]. Available on Internet: http://www.biopharminternational.com/introduction-validation-biopharmaceuticals
6 USP Validation and Verification Expert Panel (2013). Lifecycle Management of Analytical Procedures: Method Development, Procedure Performance Qualification, and Procedure Performance Verification. Stimuli to the Revision Process Article. [Accessed on 1 March 2017]. Available on Internet: https://www.usp.org/sites/default/files/usp_pdf/EN/USPNF/revisions/lifecycle_pdf.pdf
7 Shabir, G. A. (2004). Step-by-Step Analytical Methods Validation and Protocol in the Quality System Compliance Industry. IVT Network: Analytical Method Validation, pp. 4-14. [Accessed on 2 March 2017]. Available on Internet: http://www.ivtnetwork.com/sites/default/files/Analytical%20Method%20Validation.pdf
8 U.S. Food and Drug Administration. Code of Federal Regulations, Title 21, Parts 211.165(e), 211.194(a) and 211.100(a). [Accessed on 2 March 2017]. Available on Internet: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?cfrpart=211
9 International Conference on Harmonization (2005). Harmonised Tripartite Guideline: ICH Q2(R1) Validation of Analytical Procedures, Text and Methodology. [Accessed on 1 March 2017]. Available on Internet: http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html
10 McDowall, R. D. (2014). GLP and GMP Approaches to Method Validation – Going the same Way?. Spectroscopy, 29(4). [Accessed on 1 March 2017]. Available on Internet: http://www.spectroscopyonline.com/glp-and-gmp-approaches-method-validation-going-same-way
11 U.S. Pharmacopeial Convention (2016). General Chapter Prospectus: <1220> The Analytical Procedure Lifecycle. USP-NF, Notices. [Accessed on 2 March 2017]. Available on Internet: http://www.usp.org/usp-nf/notices/1220-analytical-procedure-lifecycle
12 U.S. Pharmacopeial Convention (2014). New USP requirements for Analytical Method Validation. USP-NF, Notices. [Accessed on 2 March 2017]. Available on Internet: http://www.usp.org/usp-nf/pharmacopeial-forum
13 ECA Academy (2017). Revised USP Chapter <1225> “Validation of Compendial Methods” approved. [Accessed on 2 March 2017]. Available on Internet: http://www.gmp-compliance.org/gmp-news/revised-usp-chapter-1225-validation-of-compendial-methods-approved
14 Huber, L. (2015). Recent Updates and Trends in Analytical Method Validation. PPP of The Agilent Critical Compliance Seminar. [Accessed on 7 March 2017]. Available on Internet: http://www.agilent.com/cs/library/flyers/Public/Recent_regulatory_updates_and_trends_in_analytical_method_validation.pdf
Figure 1: Huber, L. (2015). Recent Updates and Trends in Analytical Method Validation. PPP of The Agilent Critical Compliance Seminar. [Accessed on 7 March 2017]. Available on Internet: http://www.agilent.com/cs/library/fl
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal