Study of Ceramic Perovskites-type Oxides (ABO3)
| ✅ Paper Type: Free Essay | ✅ Subject: Physics |
| ✅ Wordcount: 1230 words | ✅ Published: 12 Mar 2018 |
2.1 Introduction
Ceramic perovskites-type oxides (ABO3) have been studied extensively due to the high conductivity and low activation energy. Among of perovskite type-oxide, an investigation of cerate zirconate attracts great attention to the researcher. It is well known as ion conductor and good chemical stability at intermediate temperature. It properties give big impact for development in technological applications like fuel cells, solar cells, batteries, etc. In addition, one advantages of perovskite is low cost as it can be made from common metals and industrial chemicals. According to Abdullah et al (2012), due to their low activation energy for proton conduction, the perovskite structure with proton-conducting electrolyte material important for development of Solid Oxide Fuel Cell (SOFCs) at intermediate temperature.
The investigation of proton conductivity in perovskite has started more than two decades ago. Nd doped BaCeO3 and Y doped BaZrO3 indicate good proton conducting properties under humid at elevated temperature (Azad & Irvine, 2007). Meanwhile, alkaline earth zirconates has lower proton conductivity but generally, better chemical and mechanical stability corresponding cerates (Abdullah, Hasan, & Osman, 2013).
SOFCs enable to convert chemical fuels directly into electrical power. The using SOFC used a ceramic electrolyte requires operating at high temperature and it will put this type of SOFC very great demands on the materials and technology lead to significant challenge for further development of the SOCF. Previous report has shown that the high temperature sintering resulted in large particle aggregation and growth and also consumed cost and time. Wet chemical methods (WCMs) used was able to lower the temperature as well as synthesizing time but the impurities that needed to be avoided still exist. Thus, many researchers analyze on the using of chelating agent to hinder the impurities (Abdullah, Hasan, Osman & Nordin, 2012).
2.2 Hartree-Fock
Hartree-Fock (HF) is the basis of molecular orbital (MO) Theory. HF method is an approximation method for determining the energies and wave functions in quantum mechanics. Unlike Density Functional Theory (DFT), the approximation of HF theory involves only exchange functional. It often gives qualitatively correct result. It can be systematically improved the result by carried out an MP2 or MP4 calculation, for example. HF theory was developed to solve the electronic Schrodinger equation resulted from time-dependent Schrodinger equation after refer to Bohn-Oppenheimer. The energy and many other properties of the particles can be obtained by solving Schrodinger Equation for wavefunction,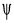 . The Schrodinger equation describes the wavefunction of a particle:
. The Schrodinger equation describes the wavefunction of a particle:
 (1)
(1)
 = wavefunction
= wavefunction
m = mass of particle
h = Planck’s constant
V = potential field in which the particle is moving
In molecular system, is a function of the positions of the electrons and the nuclei within the molecule, which will be designated as  and
and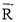 , respectively.
, respectively.
2.2.1 Molecular Hamiltonian
The Hamiltonian is made up of kinetic and potential energy. The kinetic energy is a summation of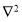 over all the particles in the molecular and the potential energy component is the Coulomb repulsion between each pair of charged entities. T:
over all the particles in the molecular and the potential energy component is the Coulomb repulsion between each pair of charged entities. T:
 (2)
(2)
 (3)
(3)
 (4)
(4)
where is the distance between two particles, and ej and ek are the charges on particles j dan k. For an electron, the charge is negative, –e while for the nucleus the charge is positive, Ze, where Z is the atomic number for that atom. The full Hamiltonian can be written as:
is the distance between two particles, and ej and ek are the charges on particles j dan k. For an electron, the charge is negative, –e while for the nucleus the charge is positive, Ze, where Z is the atomic number for that atom. The full Hamiltonian can be written as:
 (5)
(5)
From Born-Oppenheimer approximation which allows two parts of the problem to be solved independently, the kinetic energy for nuclei in Hamiltonian.
2.3 Density Functional Theory
Density functional theory (DFT) is a quantum mechanical method that be used in physics field and has become one of the most commonly used techniques in computational chemistry. DFT is a well-known quantum mechanical method to investigate complex many-body problems at the electronics structure level such as charge, bond length, density and energy. Various names for DFT models are named through combination of exchange and correlation functional.
DFT has two functional which is traditional functional and hybrid functional. The traditional functionals consist two types correlation components which are correlation functional and gradient-corrected functional. Correlation functionals involve only the values of the electron spin densities while gradient-corrected functionals involve both the values of the electron spin densities and their gradients. For the hybrid functional, it consist the combination of Hartree-Fock exchange and DFT exchange-correlation. For B3LYP, it contains the Becke Three Parameter Hybrid Functionals that using non-local correlation provided by Lee, Yang and Parr functionals, abbreviated as B3LYP. B3LYP exchange-correlation functional is:
 (6)
(6)
Where a0 = 0.20, aX = 0.72, and aC = 0.81, while the 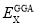 and
and  are generalized gradient approximation (GGA),
are generalized gradient approximation (GGA),  is the VWN local density approximation (LDA) to the correlation functional. GGA originally are called nonlocals approximations or semilocal approximations. The gradient of density is usually determined numerically. However, GGA has limited applicapability. It cannot describe r
is the VWN local density approximation (LDA) to the correlation functional. GGA originally are called nonlocals approximations or semilocal approximations. The gradient of density is usually determined numerically. However, GGA has limited applicapability. It cannot describe r limit of xc-energy density and the xc-potential simultaneously correctly.
limit of xc-energy density and the xc-potential simultaneously correctly.
Eschrig (1996) said the DFT method was important in providing the interested mathematician with the physicist’s view on the logical roots of the theory and also for those who want to get deeper insight into the meaning of the results of practical calculations. In addition, from previous study, DFT method was able to justify the interaction between ligands and metal fragments in coordination compound (Chermette, 1998).
2.4 Basis Set
A basis set defined as the mathematical description of the orbitals within a system used to perform the theoretical calculation. Standard basis sets for electronic structure calculations use linear combinations of Gaussian functions to create the molecular orbitals. There are few types of basis set effects such as minimal basis sets, split valence basis sets, polarized basis sets and diffuse functions basis sets. Minimal basis sets on each atom in the molecules contain minimum number of basis function and they are fixed-size atomic-type orbital. Split valence basis sets can be made larger by increase the number of basis functions of each atom. The orbital is allowed to change the size but not the shape. In contrast with polarized basis sets that allow orbital to change size and shape by adding orbitals with angular momentum beyond what is required for the ground state to the description of each atom.
The molecular orbitals are fixed linear combinations from one-electron functions and known as basis functions. They are centered on the nuclei of atom and share some similarity to atomic orbitals. An individual molecular orbital is defined as:
 (7)
(7)
The coefficients cµi are known as the molecular orbital expansion coefficients. The further explanation for the above equation can be obtained in “Exploring Chemistry with Electronic Structure Method” books.
2.6 Metal Ligand Complexes
The metal chelate has its own unique properties. The same ligand with different metal chelates share similar properties. The chelating agent complexes with the metal cation, forming a three-dimensional structure that blocks the ion’s normal reactive sites and prevents it from reacting as it normally would (“Chemical Properties of Chelates,” n.d.). The chelating agents can form coordination compounds with a metal ion as it is usually contain donor atoms like nitrogen and oxygen (Leopold et al., 2008).
One of the acti ve investigations of the synthesizing methods for the formation of a single- phase cerate zirconate powder is the lowering of the processing temperature. Using different chelating agent can lower the temperature processing. Different strength of chelating agent gives different interaction during the chelation process(Abdullah et al., 2013). A recent report shows that the temperature can be lowered by combining TETA and Ba2+ cation in forming the ligand-metal complexes solution. The chelating agent of TETA effectively reduces the formation of BaCO3 in final powder (Abdullah et al., 2012).
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal