Quantum Chemistry: Molecular Geometry of Water Molecule
| ✅ Paper Type: Free Essay | ✅ Subject: Engineering |
| ✅ Wordcount: 1099 words | ✅ Published: 30 Aug 2017 |
Question 1:
Optimize the molecular geometry of a water molecule (H2O) at HF/STO-3G level of theory in Gaussian-09 through the GaussView Visualization software package on the desktops provided.
- Give optimized bond length lengths and angles using this combination of methods and basis set.
Ans. Bond Length = 0.98927 Å and the Bond Angle H-O-H is 100.035 degrees
- Recalculate the geometry using an alternative method of your own choice.
Ans. Using Semi-empirical (PM6) we get a bond length of 0.94911 Å and a Bond Angle H-O-H is 107.488 degrees.
- Give molecular orbital diagram with drawings of the molecular orbitals. You may have to rerun the calculation with pop=full included.
Ans. Molecular orbitals for isolated H-O-H molecule were calculated using Hartree-Fock wave function and STO-3G basis set. [1][2]
- HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Occupied Molecular Orbital)
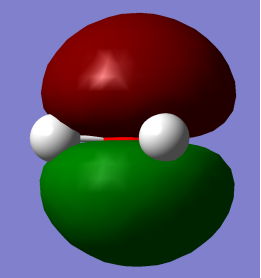

HOMO LUMO
HOMO: – Mainly pz2 is character with no contribution from Hydrogen 1s orbital but contributes to lone pair effects.
LUMO: – O-H antibonding with greatest electron density around Oxygen atom.
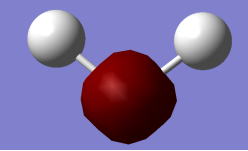 Lowest Energy Orbital 1a1 contributed by 1s orbital of Oxygen Atom (approximately spherical).
Lowest Energy Orbital 1a1 contributed by 1s orbital of Oxygen Atom (approximately spherical).
 2nd Lowest Energy Orbital 2a1 (close to non-bonding) contributed mostly by 2s orbital of Oxygen Atom (approximately spherical). Also contributes to O-H bonds.
2nd Lowest Energy Orbital 2a1 (close to non-bonding) contributed mostly by 2s orbital of Oxygen Atom (approximately spherical). Also contributes to O-H bonds.
 Energy Orbital 1b2 (non-bonding) contributed by 1s orbital of Hydrogen Atom and 2s plus 2px orbitals of Oxygen Atom leading to O-H bonds.
Energy Orbital 1b2 (non-bonding) contributed by 1s orbital of Hydrogen Atom and 2s plus 2px orbitals of Oxygen Atom leading to O-H bonds.
 Energy Orbital 3a1 (non-bonding) contributed by 1s orbital of Hydrogen Atom and 2s plus 2pz orbitals of Oxygen Atom leading to O-H bonds.
Energy Orbital 3a1 (non-bonding) contributed by 1s orbital of Hydrogen Atom and 2s plus 2pz orbitals of Oxygen Atom leading to O-H bonds.
 Highest occupied molecular orbital 1b1 (non-bonding) with pz2 character. No contribution from Hydrogen atoms.
Highest occupied molecular orbital 1b1 (non-bonding) with pz2 character. No contribution from Hydrogen atoms.
- How will the geometry change when and electron from the highest occupied molecular orbital is removed? Calculate the energy of H2O+, i.e. water with a charge of 1 and multiplicity 2.
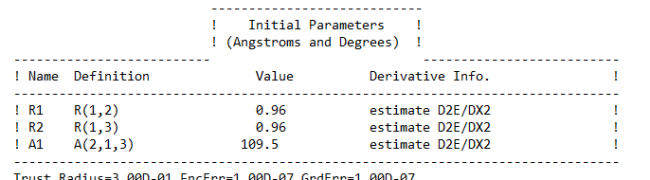
Ans. Using Hartree-Fock Wave Function with STO-3G basis set for a Water molecule with +1 charge and multiplicity 2, we get
Bond length = 0.96 Å
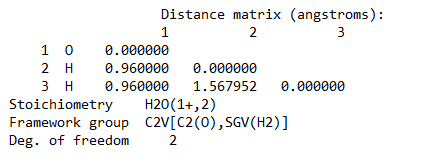
Bond Angle H-O-H = 109.5 degrees
Energy of H2O+ = -75.2017003581 A.U. (atomic units)

- How will the geometry change when an electron is removed from the second highest occupied molecular orbital of H2O?
Ans. If an electron is removed from the second highest occupied molecular orbital and electron from the highest occupied molecular orbital will move down to stabilize the oxygen atom and giving it a negative charge leading to a single lone pair. The original water molecule has 2 lone pairs and repulsion leads to a bond angle approximately 104.5 degrees, on removing an electron the repulsion force decreases leading to a larger bond angle but the geometry will remain the same.
- Calculate the infrared spectrum of water
Ans. For Hartree-Fock Wave Function and STO-3G basis set.
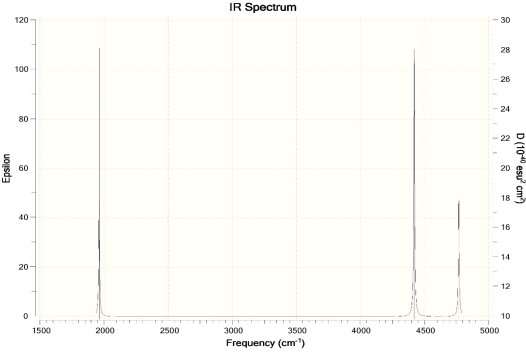
- How will the spectrum change when the hydrogen atoms are replaced by deuterium atoms?
Ans. If the hydrogen atoms were replaced by deuterium the mass of the atoms bonding to the oxygen atom increases as deuterium is a heavier isotope that results in a drop in the frequency of vibration of the molecules with similar peaks.
- How will the spectrum change when the water molecule is in liquid phase rather than in the gas-phase?
Ans. For Hartree-Fock Wave Function and STO-3G basis set.
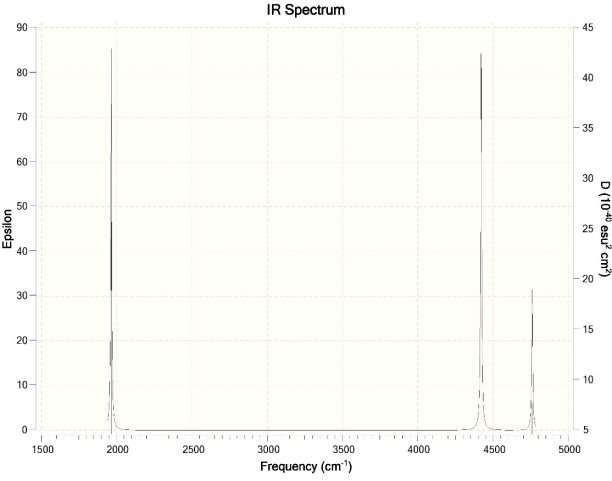
Question 2:
Do a geometry optimization and frequency for cyclohexane in the chair and boat configurations in the gas phase. Template structures should be available in GaussView. Chose a density functional method and basis set. As this is a relatively large system, I would choose a modest basis set without polarization and diffuse basis functions.
- Which of the two structures is more stable?
Ans. The chair conformer is more stable as compared to boat as the hydrogens in the chair conformation are well separated as compared to the boat conformer leading to less force of repulsion hence less energy and more stability. [3]
- Calculate the vibrational spectra of both structures. Give a comparison.
Ans. Chair Conformation IR Spectra ->
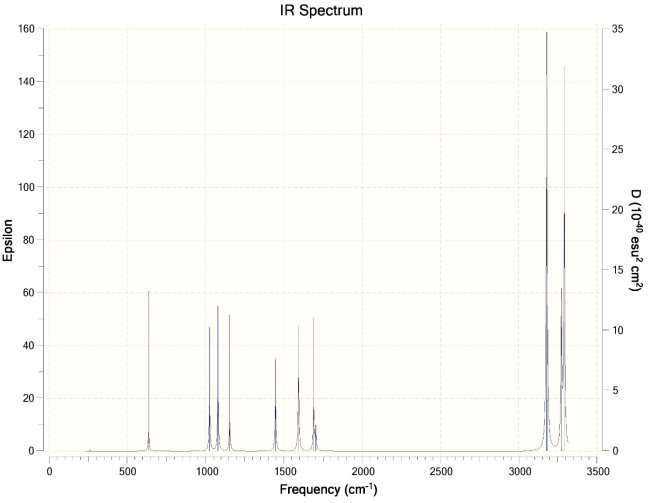
Boat Conformation IR Spectra ->
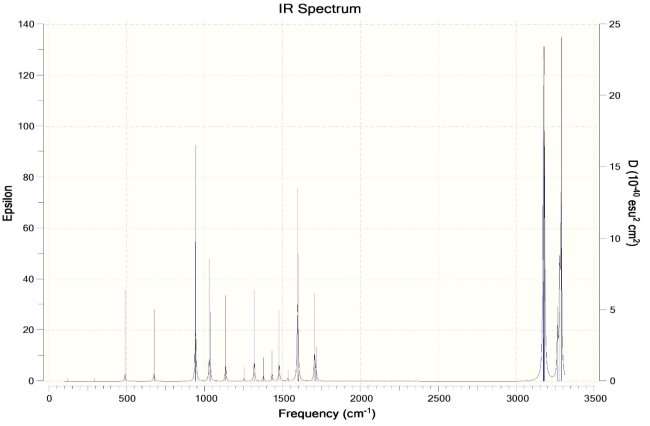
More peaks in the IR spectra of the boat conformation due to more interactions. Higher energy due to more interactions as compared to chair conformation. The boat conformation is not stable and is only used for experimental purposes and cannot exist independently.
- How would you be able to identify percentages of chair and boat configuration from a mixture of the two?
Ans. We can easily calculate the percentages of chair to boat by calculating the value of ΔG between chair and boat and equating it to (- RT ln (Q)).
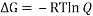
The value of Q will give us the ratio of boat to chair conformer in a mixture of two.
Chair Conformer Energy

Boat Conformer Energy

There will be a negligible difference between the energy for Chair Conformer at 10 cycles rather than 9 cycles.
Hence,
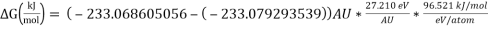 = 28.077 kJ/mol [5]
= 28.077 kJ/mol [5]
Equating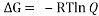 , where R= 0.008314 kJ/molK and T=273.15+27=200.15 K we get,
, where R= 0.008314 kJ/molK and T=273.15+27=200.15 K we get,
Q=1.29866 * 10^-5 which is the ratio of boat to chair conformer present in the solution.
- Draw the dipole moment of the chair and boat configuration. Which of those structures will dissolve better in water and why?
Ans. Chair conformer of cyclohexane has negligible dipole moment due to symmetry and equal charge distribution. On the other hand Boat conformation of cyclohexane has dipole moment due to the shape of the conformer making it polar due to charge distribution and steric effects.
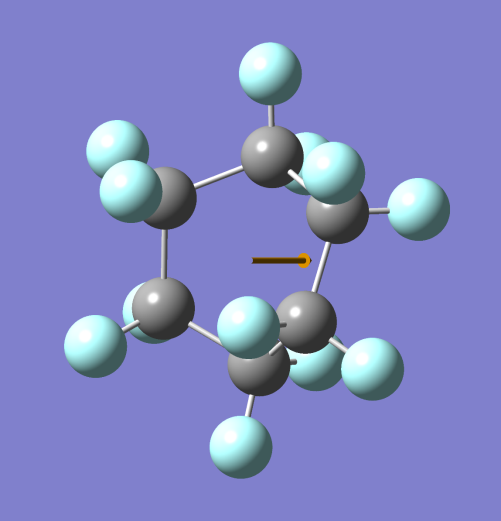
BOAT CONFORMER (Note: – Grey spheres are C atoms and Blue spheres are H atoms)
Hence the boat conformer is able to dissolve in water but stabilizes soon and turns into the chair conformer making it non-polar and separating it from water.
- How many different configurations of Fluoro-cyclohexane exist? Draw structures but do not minimize in Gaussian.
Ans. Dipole moments of Fluoro-Cyclohexane have increased by a factor of 611.6 as compared to cyclohexane.


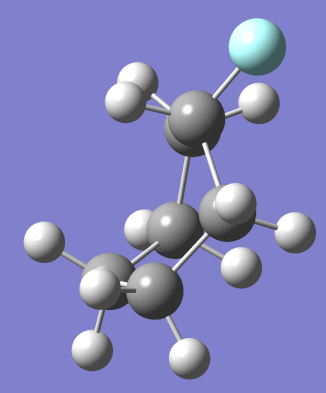 This explains how the presence of a single fluoride atom instead of a hydrogen impacts the structure and charge distribution of the cyclic hydrocarbon. In turn adding a Fluoride atom also increases the energy of the cyclic hydrocarbon creating less stable structures in boat, twist and half chair increasing
This explains how the presence of a single fluoride atom instead of a hydrogen impacts the structure and charge distribution of the cyclic hydrocarbon. In turn adding a Fluoride atom also increases the energy of the cyclic hydrocarbon creating less stable structures in boat, twist and half chair increasing  and decreasing the amount of other conformers in a solution compared to chair. [4]
and decreasing the amount of other conformers in a solution compared to chair. [4]
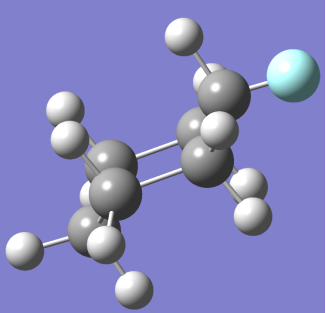
CHAIR CONFORMER BOAT CONFORMER
(NOTE: – Grey spheres are C atoms, White Spheres are H atoms and Blue Sphere is F atom)
At room temperature only Chair Fluoro-Cyclohexane can exist but may transition between conformations that will be present for negligible time.
REFERENCES:
SOFTWARES:
- Gaussian(R) 09
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal