Pain Induced Synaptic Plasticity in the Amygdala
Info: 45772 words (183 pages) Dissertation
Published: 24th Feb 2022
Tagged: BiologyBiomedical Science
Abstract
Pain is an important defense against dangers in our environment, however some clinical conditions produce pain that outlasts this useful role and persist even after the injury has healed. The experience of pain consists of somatosensory elements of intensity and location, negative emotional/aversive feelings and subsequent restrictions on lifestyle due to learning to associate certain activities with pain. The amygdala contributes negative emotional value to nociceptive sensory information and forms the association between an aversive response and the environment in which it occurs. It can form this association because it receives nociceptive information via the spino-parabrachio-amygdaloid pathway and polymodal sensory information via its basolateral nucleus (BLA) and cortical and thalamic inputs. Within the spino-parabrachio-amygdaloid pathway, nociceptive information is sent from the external lateral nucleus of the parabrachial nucleus (PB) to the laterocapsular region of the central nucleus of the amygdala (CeLC). The PB-CeLC synapse and other brain regions undergo synaptic plasticity in chronic pain conditions with ongoing injury. However very little is known about how plasticity occurs in conditions where pain persists even after the injury has healed.
In the first study of this thesis, I used immunohistochemistry, electrophysiology and behavioural assays, to show that a brief nociceptive stimulus with no ongoing injury can produce long-lasting synaptic plasticity at the rat parabrachial-amygdala synapse. I show that this plasticity is caused by an increase in postsynaptic AMPARs with a transient change in AMPAR subunit, similar to long-term potentiation. Furthermore, repeated stimuli lengthened this plasticity. The potentiation could be representative of the initial changes that occur in the transition from an acute to a chronic pain state. This could mean greater association of a painful experience with the environment and context and could ultimately facilitate the negative association of certain activities and situations with pain, leading to limiting or avoidance of these activities/situations.
The next studies of this thesis focused on potential neuromodulators of activity at the PB-CeLC synapse and the polymodal BLA inputs to the CeLC. Opioids and Calcitonin gene-related peptide (CGRP) were chosen because of their presence at synapses in the amygdala and their role in pain particularly in the affective component of pain. Opioids reduce pain intensity and the emotional unpleasantness of pain. Opioids inhibit some synapses in the amygdala, however whether opioids specifically modulate the PB-CeLC and the BLA-CeLC is unknown. I used electrophysiology and opotogenetics to show that opioids inhibit two synapses important for pain modulation in the amygdala. Given the evidence of the opioid’s role in reducing pain affect, modulation of these synapses could be, in part, the site of opioid action. CGRP is expressed at all levels of the spino-parabrachio-amygdala pathway and modulates pain, as CGRP receptor antagonists injected into the amygdala inhibit nocifensive behaviours in animals. Additionally, CGRP antagonists reverse arthritis-induced synaptic plasticity at the PB-CeLC synapse. CGRP enhances synaptic transmission in the CeLC, however it is unknown whether it also directly regulates the excitability of CeLC neurons. Using electrophysiology, I show that CGRP directly ‘excites’ CeLC neurons even when fast synaptic transmission is blocked. This suggests that in normal physiology CGRP and opioids have opposing effects in the CeLC and the balance of activity in the CeLC will depend on which peptide has the bigger influence on the CeLC.
This thesis addressed the question of whether plasticity can outlast a stimulus and the time course of the plasticity. This plasticity was seen in the amygdala, an area important for associative learning and the affective component of pain. This thesis also addressed how neuropeptides, opioids and CGRP regulate amygdala synapses in normal physiology. Knowledge of how these peptides modulate the amygdala synapses will provide information on how they could operate in a pain state.
Abbreviations
AAV: Adeno-associated virus
ACo: Anterior cortical nuclei of the amygdala
ACC: Anterior cingulate cortex
ACSF: Artificial cerebrospinal fluid
AMPA: Alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
AMPAR: AMPA receptor
ANOVA: Analysis of variance
ATP: Adenosine triphosphate
BIB: BIBN4096BS
BLA: Basolateral nucleus of the amygdala
BLV: Basolateral ventral nuclei of the amygdala
BM: Basomedial nuclei of the amygdala
BSA: Bovine serum albumin
cAMP: Cyclic adenosine monophosphate
CAMKII: Calcium calmodulin-dependent kinase II
CeA: Central nucleus of the amygdala
CeL: Lateral region of the central nucleus of the amygdala
CeLC: Laterocapsular region of the central nucleus of the amygdala
CeM: Medial region of the central nucleus of the amygdala
CGP: CGP 55845
CGRP: Calcitonin gene-related peptide
CGRP 8-37: Calcitonin gene-related peptide fragment 8-37
ChR2: Channelrhodopsin-2
CPA: Conditioned place aversion
CRLR: Calcitonin-receptor like receptor
CS: Conditioned stimulus
DAMGO: [D-Ala2, NMe-Phe4, Gly-ol5]-enkephalin
DL-APV: DL-2-amino-5-phosphonopentanoic acid
DOR: Delta opioid receptor
EPSC: Excitatory postsynaptic current
eEPSC: Evoked EPSC
EYFP: Enhanced yellow fluorescent protein
GABA: Gamma-aminobutyric acid
GDP: Guanosine diphosphate
GFP: Green fluorescent protein
GIRK: G-protein activated inwardly rectifying potassium channel
GTP: Guanosine triphosphate
HCN: Hyperpolarisation-activated cyclic nucleotide-gated channels
IC: Insular cortex
ICI: ICI-174,864
Ih: Hyperpolarisation activated cation channel
ITC: Intercalated cells of the amygdala
I/V: current/voltage
Kir: Inwardly rectifying potassium channels
KOR: Kappa opioid receptor
LA: lateral amygdala
LTP: Long term potentiation
LV: Lentivirus
MeA: Medial nucleus of the amygdala
Met-Enk: Methoinine-Enkephalin
MOR: Mu opioid receptor
NGS: Normal goat serum
NMDA: N-methyl-D-aspartate
NMDAR: NMDA receptor
NpHR: Natronomonas pharanos halorhodopsin
Nor-BNI: Nor-Binaltorphimine
PAG: Periaqueductal gray
PB: Parabrachial nucleus
PBel: External lateral PB
PBS: Phosphate buffered saline
PFC: Prefrontal cortex
PKA: Protein kinase A
PPR: Paired pulse ratio
PTX: Pertussis toxin
PWT: Paw withdrawal threshold
RAMP: Receptor activity modifying protein
RCP: Receptor component protein
RVM: Rostral ventral medulla
SCP: Superior cerebellar peduncle
S.E.M: Standard error of the mean
SI: Somatosensory cortex
Sld: Substantia innominata dorsalis
TTL: Transistor-transistor logic
U69: U-69593
US: Unconditioned stimulus
VGCC: Voltage-gated calcium channels
VTA: Ventral tegmental area
Table of Contents
Click to expand Table of Contents
Chapter 1: Introduction
1.1 Components of Pain
1.1.1 Ascending Pain Pathways
1.1.2 Amygdala and the Descending analgesic pathways
1.2 Amygdala
1.2.1 Structure
1.2.2 Connectivity and function
1.2.3 Role of the amygdala in pain
1.3 Central sensitization and Synaptic Plasticity
1.3.1 Postsynaptic glutamate receptors
1.3.2 Pain related synaptic plasticity in the Amygdala
1.4 Opioids
1.4.1 Opioid receptors and Ligands
1.4.2 Opioid Analgesia
1.4.3 Opioid receptor effectors
1.4.4. Opioid expression in the Parabrachial nucleus
1.4.5 Opioid expression in the Amygdala
1.4.6 Opioid activity in the PB and amygdala
1.5 Calcitonin gene-related peptide (CGRP)
1.5.1 CGRP expression in the PB-CeLC synapse
1.5.2 CGRP receptor signaling
1. 5. 3 CGRP and pain
1.6 Optogenetics
1.6.1 Channelrhodopsin-2
1.6.2 Inhibitory optogenetics control
1.6.3 Viral delivery
1.6.4 Limitations of optogenetics
1.7 Summary and study aims
Chapter 2: Methods
2.1 General Methods
2.1.1 Animals
2.1.2 Preparation of brain slices
2.2.3 Data Analysis
2.2 Chapter 3 Methods
2.2.1 Nociceptive stimulus
2.2.2 Peripheral inflammation
2.2.3 Immunohistochemistry
2.2.4 Electrophysiology
2.2.5 Behavioural testing
2.2.6 Drugs
2.2.7 Data Analysis
2.3 Chapter 4 Methods
2.3.1 Electrophysiology
2.3.2 Drugs
2.3.3 Data Analysis
2.4 Chapter 5 Methods
2.4.1 Stereotaxic surgeries
2.4.2 Preparation of brain slices
2.4.3 Electrophysiology
2.4.4 Immunohistochemistry
2.4.5 Drugs
2.4.6 Data analysis
2.5 Chapter 6 methods
2.5.1 Electrophysiology
2.5.2 Drugs
2.5.3 Data analysis
Chapter 3: Central sensitization of the spino-parabrachial-amygdala pathway that outlasts a brief nociceptive stimulus
3.1 Introduction
3.2 Aims
3.3 Results
3.3.1 A brief nociceptive stimulus
3.3.2 Nociceptive stimulus induces long-lasting synaptic plasticity specifically at PB-CeLC synapse
3.3.3 Nociceptive stimulus produces a transient change in AMPAR subunit composition at PB-CeLC synapse
3.3.4 PB-CeLC synapse undergoes metaplastic-like changes
3.3.5 Nociceptive stimulus produces mechanical but not thermal hyperalgesia
3.4 Discussion
Chapter 4: Opioids differentially modulate two synapses important for pain signaling in the amygdala
4.1 Introduction
4.2 Aims
4.3 Results
4.3.1 Met-Enk inhibits the PB-CeLC synapse through a presynaptic mechanism
4.3.2 Met-enk acts on the MOR and DORs to inhibit the PB-CeLC synapse
4.3.3 Met-Enk inhibits the BLA-CeLC synapse through presynaptic MOR
4.3.4 KOR modulates the BLA-CeLC synapse
4.4 Discussion
Chapter 5: Optogenetic dissection of opioid regulation at the PB-CeLC synapse
5.1 Introduction
5.2 Aims
5.3 Results
5.3.1 Injection spread and reliability
5.3.2 Optogenetics can be used to selectively activate the PB-CeLC synapse
5.3.3 MOR, not the DOR inhibits presynaptic glutamatergic release at the PB-CeLC synapse in ChR2 animals
5.3.4 AAV5 driven expression of ChR2 alters the PPR at the PB-CeLC synapse
5.4 Discussion
Chapter 6: Calcitonin gene-related peptide as a neuromodulator of the PB-CeLC synapse
6.2 Aims
6.3 Results
6.3.1 CGRP enhances synaptic transmission at the PB-CeLC synapse through a post-synaptic mechanism
6.3.2 CGRP can directly regulate CeLC neurons
6.3.3 CGRP does not activate hyperpolarisation-activated inward currents (Ih)
6.3.4 CGRP does not activate G protein-gated inwardly rectifying potassium (GIRK) channels
6.4 Discussion
General discussion and conclusions
7.1 Opioids and CGRP in the amygdala
7.2 Mechanisms of pain induced plasticity
7.3 CeLC as the integrative center of the amygdala
7.4 Future directions and conclusion
References
List of figures
Figure 1: Schematic of the main nuclei in the amygdala
Figure 2: NMDAR dependent LTP
Figure 3: AMPA/NMDA ratio
Figure 4: A brief nociceptive stimulus without inflammation or ongoing activation of the spino-parabrachio-amygdaloid pathway
Figure 5: Measurement of AMPA/NMDA ratio
Figure 6: A brief nociceptive stimulus induces long lasting synaptic plasticity specifically at PB-CeLC synapse
Figure 7: A brief nociceptive stimulus produces a transient change in AMPAR subunit composition at PB-CeLC synapse
Figure 8: PB-CeLC synapse undergoes metaplastic-like changes following synaptic plasticity
Figure 9: Nociceptive stimulus produces mechanical but not thermal hyperalgesia
Figure 10: Met-Enk inhibits the PB-CeLC synapse through a presynaptic mechanism
Figure 11: Met-Enk acts on MOR and DORs to inhibit the PB-CeLC synapse
Figure 12: Met-Enk inhibits the BLA-CeLC synapse through presynaptic MOR
Figure 13: KOR modulates the BLA-CeLC synapse
Figure 14: AAV5 injection into the PBel produces ChR2/EYFP expression in the PBel, which can be activated by blue light
Figure 15: AAV5 injection into the PBel produces ChR2/EYFP terminal expression in the CeLC, which can be activated by light
Figure 16: MOR, not the DOR inhibits presynaptic glutamate release at the PB-CeLC synapse in ChR2 animals
Figure 17: AAV5 driven expression of ChR2 alters the PPR ratio at the PB-CeLC synapse
Figure 18: CGRP enhances synaptic transmission at the PB-CeLC synapse through a post-synaptic mechanism
Figure 19: CGRP increases excitability of CeLC neurons in the presence of fast synaptic transmission
Figure 20: CGRP can modulate CeLC neuronal excitability in the absence of fast synaptic transmission
Figure 21: CGRP, Forskolin and Met-Enk do not modulate Ih channels in CeLC neurons
Figure 22: CGRP does not activate G protein-gated inwardly rectifying potassium (GIRK) channels
Chapter 1: Introduction
Pain is an important protective mechanism against dangers in our environment. Acute pain prevents potential physical damage. However, in some cases pain outlasts this protective function and becomes chronic. Chronic pain affects 1 in 5 adults worldwide and has a significant impact on suffers (Gureje et al., 1998). Individuals with chronic pain have diminished quality of life and increased incidence of anxiety and depression (Gureje et al., 1998). In Australia, chronic pain is estimated to affect 17 percent of males and 20 percent of females (Blyth et al., 2001) and has significant economic and health costs (Phillips, 2009). Chronic pain conditions may arise from ongoing peripheral injury such as osteoarthritis and neuropathic pain or may be spontaneous in nature with no ongoing peripheral injury such as chronic back pain. Back pain is one of the most common reasons for healthcare visits and the most common anatomical pain site (Atkinson, 2004, Gureje et al., 1998). Conditions where there is no peripheral injury may be due to development of central sensitization in CNS pain related areas (Latremoliere and Woolf, 2009, Ji et al., 2003, Woolf, 2011). In such cases, initial activation of peripheral nociceptors causes CNS plasticity that outlasts the initial stimulus (Woolf, 2011, Latremoliere and Woolf, 2009, Ji et al., 2003). A better understanding of the cellular mechanism and timing of this plasticity may lead to more effective therapeutic targets for spontaneous chronic pain. The following provides a brief overview on pain transmission with a focus on the role of the amygdala in pain, how synaptic plasticity can lead to central sensitization and the neuromodulators that may regulate activity in the amygdala.
1.1 Components of Pain
The experience of pain consists of sensory-discriminative components of intensity and location and an affective/emotional component. The latter consists of the emotional unpleasantness and aversiveness of the pain experience and the avoidance behaviour that comes from the learned association of pain with certain activities. This experience is mediated by neural pain pathways that work in parallel and in series with each other to produce a behavioral response (Basbaum et al., 2009, Bushnell et al., 2013, Elman and Borsook, 2016, Kuner, 2010, Vlaeyen, 2015, Price, 2000).
1.1.1 Ascending Pain Pathways
The ascending pain pathway begins at peripheral nociceptors, which detect different noxious stimuli such as thermal, mechanical and chemical stimuli (Almeida et al., 2004, Basbaum et al., 2009, Kuner, 2010). The nociceptors transmit signals to the superficial spinal cord dorsal horn, which in turn projects to higher brain regions to produce the perception of pain (Almeida et al., 2004, Basbaum et al., 2009, Kuner, 2010). The different aspects of pain are mediated by spinal cord projections to specific targets such as the thalamus and parabrachial nucleus (Almeida et al., 2004, Basbaum et al., 2009, Kuner, 2010). The lateral thalamus (ventroposterior lateral nucleus) projects to the somatosensory cortex (SI) (Gingold et al., 1991, Almeida et al., 2004) and mediates the sensory-discriminative aspect of pain (Almeida et al., 2004, Basbaum et al., 2009, Kuner, 2010). Both the lateral thalamus and SI have small receptive fields and graded increases in pain intensity results in greater activation of both regions (Kenshalo et al., 1988, Kenshalo et al., 2000, Guilbaud et al., 1980, Peschanski et al., 1980, Zhang et al., 2011b), allowing them to sense changes in pain location and intensity. The medial thalamus projects to areas important for pain affect such as the anterior cingulate cortex (ACC) (Vogt et al., 1979), prefrontal cortex (PFC) and the insular cortex (IC) (Bornhovd et al., 2002, Buchel et al., 2002, Bushnell and Duncan, 1989, Price, 2000, Almeida et al., 2004). The ACC is the most important region in the regulation of pain affect. Suggestions of its involvement in pain affect first came from a study which found that patients who have had cingulotomy detect pain but find it less distressing (Foltz and White, 1962). Moreover, a human imaging study showed that amongst the ACC, SI and IC, only the ACC is activated when patients feel increased pain unpleasantness (Rainville et al., 1997). The ACC’s involvement in pain affect is now substantiated by animal behavioural paradigms such as conditioned place aversion (CPA) studies (Ren et al., 2006, Gao et al., 2004, Johansen et al., 2001). CPA assesses the aversiveness of the affective component of pain (Zhang et al., 2011a) and lesion or inactivation of the ACC suppresses pain-induced CPA (Ren et al., 2006, Gao et al., 2004, Johansen et al., 2001). Another important pathway for pain affect is the spino-parabrachio-amygdaloid pathway. This pathway sends purely nociceptive information from the external lateral portion of the parabrachial nucleus (PB) to the laterocapsular region of the central nucleus of the amygdala (CeLC) (Bernard et al., 1993, Bernard et al., 1992, Bester et al., 1997). Like the ACC, inactivation or lesion of the amygdala reduces pain induced CPA (Gao et al., 2004, Pedersen et al., 2007, Ansah et al., 2010, Tanimoto et al., 2003), and it associates the negative aspects of pain with the environment in which it occurs (Han et al., 2015, Sato et al., 2015). Therefore, the parabrachial-amygdala synapse is the focus of this thesis and will be discussed in greater detail in the upcoming sections.
1.1.2 Amygdala and the Descending analgesic pathways
Pain signals can be inhibited in the spinal cord before reaching the brain (Heinricher et al., 2009, Lau and Vaughan, 2014). One of the ways, this is achieved is through activation of the descending analgesic pathway. There are several regions involved in the descending modulation of pain including the periaqueductal gray-rostral ventral medulla (PAG-RVM) pathway, the dorsal reticular nucleus and caudal lateral ventrolateral medulla (Heinricher et al., 2009), with the PAG-RVM being the best understood (Millan, 2002, Heinricher et al., 2009). The amygdala sends projections to the PAG (Haubensak et al., 2010) and is also part of the PAG-RVM analgesic pathway (Manning and Meyer, 1995, Borszcz, 1999, Calvino et al., 1982). Within the PAG-RVM pathway, the PAG sends excitatory projections to the RVM, which in turn projects via the dorsolateral funiculus to the spinal cord, where it inhibits pain signals through opioid receptor activation (Heinricher et al., 2009, Millan, 2002, Lau and Vaughan, 2014).
1.2 Amygdala
1.2.1 Structure
The amygdala is an almond shaped structure located in the temporal lobe of the brain and is thought to provide emotional value to sensory information (Sah et al., 2003, Veinante et al., 2013, Neugebauer, 2015, LeDoux, 2000). It is composed of several diverse nuclei, which are subdivided into 4 main groups (Veinante et al., 2013, Sah et al., 2003) (Figure 1). The basolateral group consists of the lateral nucleus (LA), the basolateral nucleus (BLA) and the accessory basal nuclei (basolateral ventral and basomedial nuclei). The central nucleus is further divided into the laterocapsular nucleus (CeLC), the lateral nucleus (CeL) and the medial nucleus (CeM). The other two groups are the medial nucleus (MeA) and the superficial group. The intercalated cell masses are not part of this classification, but they also have a prominent role in amygdala function (Veinante et al., 2013, Sah et al., 2003) (Figure 1). The BLA and the CeA specifically the CeLC are the nuclei involved in pain and hence they will be the focus of this study.
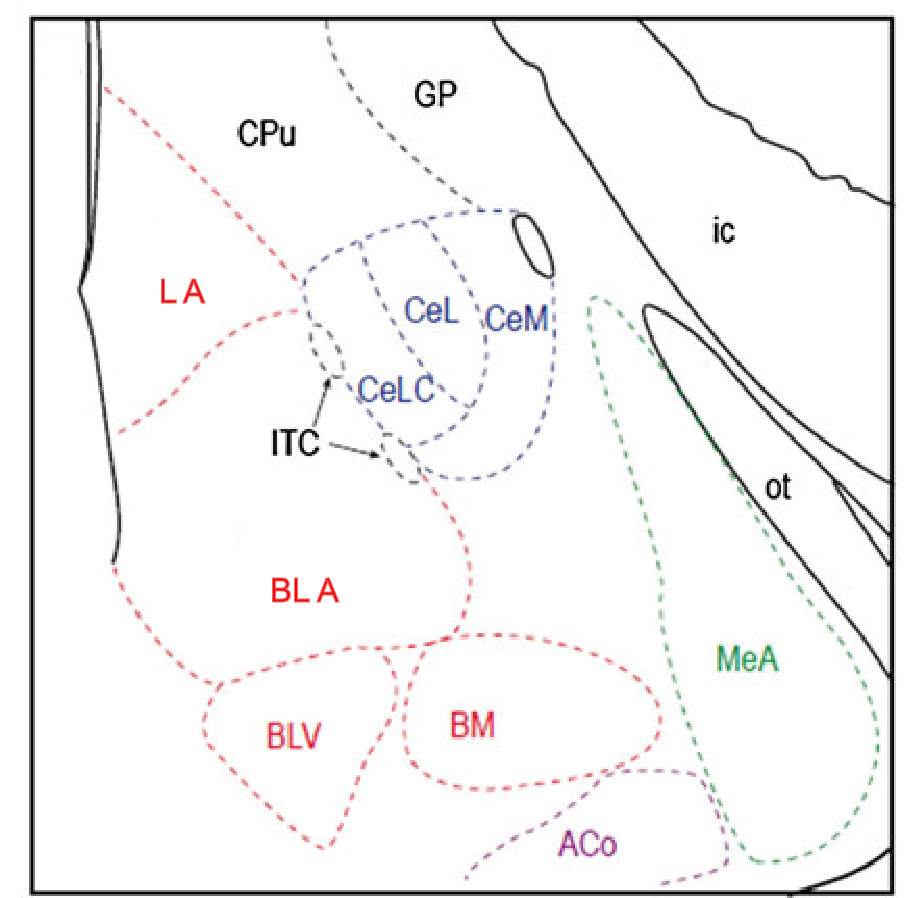
Figure 1: Schematic of the main nuclei in the amygdala
The basolateral group (red) consists of the Lateral (LA), basolateral (BLA) and basolateral ventral (BLV) and basomedial nuclei (BM). The central nucleus (blue) includes the laterocapsular (CeLC), lateral (CeL) and medial (CeM) nucleus. The medial group (green) and superficial (purple) are represented at this bregma by the medial (MeA) and anterior cortical (ACo) nuclei respectively. The intercalated cells (ITC) are found between the CeA and the BLA. Other abbreviations: CPu: caudate putamen, ic: internal capsule, GP: globus pallidus and ot: optic tract. Image modified from Veinate et al 2013.
1.2.2 Connectivity and function
The CeLC is the amygdala’s main source of nociceptive information. It receives specific nociceptive information from the external lateral PB (Bernard et al., 1993, Bernard et al., 1992, Bester et al., 1997) and highly processed polymodal sensory information including nociceptive information from the BLA. The BLA receives nociceptive information from the thalamus (Moga et al., 1995, Vertes and Hoover, 2008, Turner and Herkenham, 1991) and other sensory information from the lateral orbital area (McDonald et al., 1996), auditory thalamus (LeDoux et al., 1990b, Turner and Herkenham, 1991, Doron and Ledoux, 1999) and auditory cortex (Romanski et al., 1993). It also receives inputs from higher order cortical regions such as the entorhinal cortex (McDonald and Mascagni, 1997), prelimbic cortex, ACC and IC (McDonald et al., 1996). These inputs project from the BLA onto the CeLC (Pitkanen et al., 1997, Sah et al., 2003, Neugebauer, 2015), providing it with polymodal sensory information that has already undergone significant processing. Further to this, the CeLC itself receives sensory inputs from thalamus (Moga et al., 1995, Vertes and Hoover, 2008), hypothalamus (Canteras et al., 1994), entorhinal cortex (McDonald and Mascagni, 1997) and lateral occipital area (McDonald et al., 1996) and inputs from areas delivering pain affect information such as prefrontal cortex, IC and ACC (McDonald et al., 1996). Thus, given the sensory and affective information it receives, the CeLC may also be important for associating the negative aspects of pain with the environment in which it occurs.
1.2.3 Role of the amygdala in pain
The amygdala is an important component of the pain matrix. It is activated in acute and chronic pain states (Bornhovd et al., 2002, Baliki et al., 2006) and receives nociceptive information via the PB-CeLC synapse (Bernard et al., 1993, Bernard et al., 1992, Bester et al., 1997) and the BLA-CeLC synapse (Sah et al., 2003, Marek et al., 2013, Pape and Pare, 2010, Neugebauer, 2015). Inactivation of the BLA (Hebert et al., 1999, Ji et al., 2010) and CeA (including CeLC) (Hebert et al., 1999, Han and Neugebauer, 2005, Han et al., 2005, Carrasquillo and Gereau, 2007, Pedersen et al., 2007, Fu and Neugebauer, 2008, Ji et al., 2010) reduces nocifensive behaviour in animals. Conversely, direct pharmacological activation of the CeLC, the nucleus that receives nociceptive information increases pain hypersensitivity in the absence of tissue damage (Han et al., 2010, Kolber et al., 2010, Carrasquillo and Gereau, 2007, Ji et al., 2013, Li et al., 2011).
The amygdala isimportant for emotional processing (Kluver and Bucy, 1997, Adolphs et al., 1994, Young et al., 1995, Bechara et al., 1995, LeDoux, 2000, Cardinal et al., 2002). In humans, damage to the amygdala impairs recognition of emotions in facial expressions (Adolphs et al., 1994, Young et al., 1995) and the ability to form the association between a conditioned and unconditioned stimulus (Bechara et al., 1995). In monkeys removal of the temporal lobe leads to complete absence of emotional reactions, particularly fear (Kluver and Bucy, 1997). Given the importance of the amygdala in emotional responses this raised the possibility that the amygdala may participate in the affective/emotional experience of pain. Several lines of evidence now support this proposition. Firstly, CeA neurons have a sigmoid stimulus-response curve (Bernard et al., 1990, Bernard et al., 1992, Neugebauer and Li, 2002). This means an increase in nociceptive stimulation initially leads to increase in activation of CeA neurons. The increase in activation then reaches a plateau, where subsequent increases in nociceptive stimulation does not change CeA neuronal activation. This, combined with their large bilateral receptive fields, makes them ineffective for sensory-discrimination of pain (Bernard et al., 1990, Bernard et al., 1992, Neugebauer and Li, 2002). Furthermore, inactivation of the CeA’s source of nociceptive input, the external lateral PB attenuates affective pain behaviour such as escape following foot shock (Han et al., 2015). As well as pain, the BLA and CeA are also involved in fear learning/conditioning and hence associative learning (Cardinal et al., 2002, Sah et al., 2003, Pitkanen et al., 1997, LeDoux, 2000). This raises the possibility that they have the same role in the affective component of pain. Fear as an emotion is similar to pain, in that it has a protective function. Fear leads to adaptive behaviours such as avoidance of a dangerous environment or situation. This is similar to the affective component of pain where association of pain with pain inducing activities leads sufferer’s to limit those activities (Vlaeyen, 2015). The experimental paradigm of fear, Pavlovian fear conditioning/learning involves the association between an aversive unconditioned stimulus (US) with a neutral conditioned stimulus (CS) to produce defensive or escape behaviours (Arruda-Carvalho and Clem, 2015, LeDoux, 2000, Cardinal et al., 2002). This association occurs within the LA-BLA network (LeDoux et al., 1990a, Campeau and Davis, 1995, Helmstetter and Bellgowan, 1994, Arruda-Carvalho and Clem, 2015) which then acts on the CeA (Campeau and Davis, 1995, Hitchcock and Davis, 1986, Cardinal et al., 2002), whose outputs modify fear behaviours such as freezing (LeDoux et al., 1988, Cardinal et al., 2002). Most fear-learning studies implicate the LA-BLA network (LeDoux, 2000, Arruda-Carvalho and Clem, 2015) but recent studies have shown that the CeA can also mediate fear learning in parallel to LA-BLA (Ciocchi et al., 2010, Li et al., 2013, Haubensak et al., 2010). Of particular interest is the recent evidence that the PB-CeLC synapse is also involved in fear learning (Sato et al., 2015, Han et al., 2015, Watabe et al., 2013). Activation of this synapse can induce a fear response, in the absence of the typically used US foot shock. Conversely inactivation of this synapse can attenuate the acquisition of fear (Han et al., 2015, Sato et al., 2015). This type of associative learning in response to pain is demonstrated in CPA studies. Similar to Pavlovian fear conditioning/learning, CPA requires the animal to associate the pain experience with the context to formulate adaptive behavioural responses, in this case avoidance of the environment where the pain was experienced. Lesion or inactivation of the CeA suppresses pain induced CPA (Gao et al., 2004, Pedersen et al., 2007, Ansah et al., 2010, Tanimoto et al., 2003). Thus, the CeA associates the negative aspects of pain with the environment in which it occurs, leading to avoidance behaviours in pain sufferers. The CeA can also be antinociceptive (Manning and Meyer, 1995, Borszcz, 1999, Calvino et al., 1982). Lesions of the CeA reduce the antinociceptive effect of morphine injected into the PAG (Manning and Meyer, 1995, Borszcz, 1999, Calvino et al., 1982), suggesting that the CeA projections to the PAG (Haubensak et al., 2010) are part of the descending analgesic pathway.
The amygdala is important in the regulation of pain. The CeLC through its nociceptive and polymodal inputs is important for the associative learning involved in the affective component of pain and is also part of the descending analgesic pain pathway. The next section will look at synaptic plasticity and how it leads to central sensitization in pain pathways and how synaptic plasticity in the amygdala, particularly at the PB-CeLC synapse, affects pain behaviour.
1.3 Central sensitization and Synaptic Plasticity
Central sensitization is the enhancement of the neural circuits responsible for pain in response to a nociceptive stimulus and results from synaptic plasticity of these circuits (Basbaum et al., 2009, Ji et al., 2003, Latremoliere and Woolf, 2009, Woolf, 2011). This synaptic plasticity enhances the nocifensive response to nociceptive stimuli (Latremoliere and Woolf, 2009, Woolf, 2011) and can result in a nocifensive response independently of peripheral activation of nociceptors (Latremoliere and Woolf, 2009, Woolf, 2011). Central sensitization increases pain hypersensitivity by changing receptive fields, causing pain outside the site of injury (secondary hyperalgesia) (Latremoliere and Woolf, 2009, Woolf, 2011, Cook et al., 1987, Woolf, 1983), reducing pain thresholds, making previously innocuous stimuli noxious (allodynia) and heightening the response to already noxious stimuli (hyperalgesia) (Basbaum et al., 2009, Ji et al., 2003, Latremoliere and Woolf, 2009, Woolf, 2011).
Synaptic plasticity is the ability of synapses to change their efficacy and strength in response to activity (Chater and Goda, 2014, Citri and Malenka, 2008, Collingridge et al., 2004, Isaac et al., 2007). Synaptic plasticity is critical for normal function particularly memory formation (Chater and Goda, 2014, Citri and Malenka, 2008, Collingridge et al., 2004, Isaac et al., 2007) and as mentioned is the underlying mechanism of central sensitization (Basbaum et al., 2009, Ji et al., 2003, Latremoliere and Woolf, 2009, Woolf, 2011, Luo et al., 2014). The mechanism of synaptic plasticity in central sensitization is commonly changes in postsynaptic glutamate receptors (Latremoliere and Woolf, 2009, Ji et al., 2003). The next section will provide an overview on postsynaptic mechanisms of synaptic plasticity, the evidence for pain-related synaptic plasticity in the amygdala and its influence on pain behaviour.
1.3.1 Postsynaptic glutamate receptors
Long-term potentiation (LTP) is an activity dependent form of synaptic plasticity that changes postsynaptic glutamate receptors (Chater and Goda, 2014, Citri and Malenka, 2008, Collingridge et al., 2004, Isaac et al., 2007). LTP is the best-studied form of synaptic plasticity and is a synaptic model of learning and memory. In classical LTP, brief high intensity activity activates AMPARs, which depolarizes the cell, relieving the voltage dependent magnesium block from NMDARs (Chater and Goda, 2014, Citri and Malenka, 2008, Collingridge et al., 2004, Isaac et al., 2007). NMDARs, because of their calcium permeability, can initiate several calcium dependent-signaling pathways that result in the incorporation of AMPARs at the postsynaptic density (Figure 2) (Chater and Goda, 2014, Citri and Malenka, 2008, Collingridge et al., 2004, Isaac et al., 2007). One of these calcium dependent pathways is activation of CAMKII (Figure 2) (Malenka and Nicoll, 1999, Kauer and Malenka, 2007). CAMKII is part of a family of calmodulin (CaM) kinases that phosphorylates the serine/threonine residues of their protein substrates in order to change their function (Wayman et al., 2008). In some forms of LTP, the incorporation of AMPARs may involve the transient incorporation of calcium permeable GluA2-lacking AMPARs, which are eventually replaced with calcium impermeable GluA2-containing AMPARs (Morita et al., 2014, Plant et al., 2006, Guire et al., 2008). GluA2-lacking AMPARs can facilitate synaptic plasticity independent of NMDARs due to their calcium permeability (Guire et al., 2008, Asrar et al., 2009, Whitehead et al., 2013). Stress (Whitehead et al., 2013), stroke (Quintana et al., 2015), sensory deprivation (Takahashi et al., 2003) and pain (Chen et al., 2014, Cheng et al., 2011) increase the number of postsynaptic AMPARs at synapses, whilst the number of NMDARs remains unchanged. These changes in relative glutamate receptor number increase the AMPA/NMDA ratio of the EPSC (Figure 3) (Kauer and Malenka, 2007).
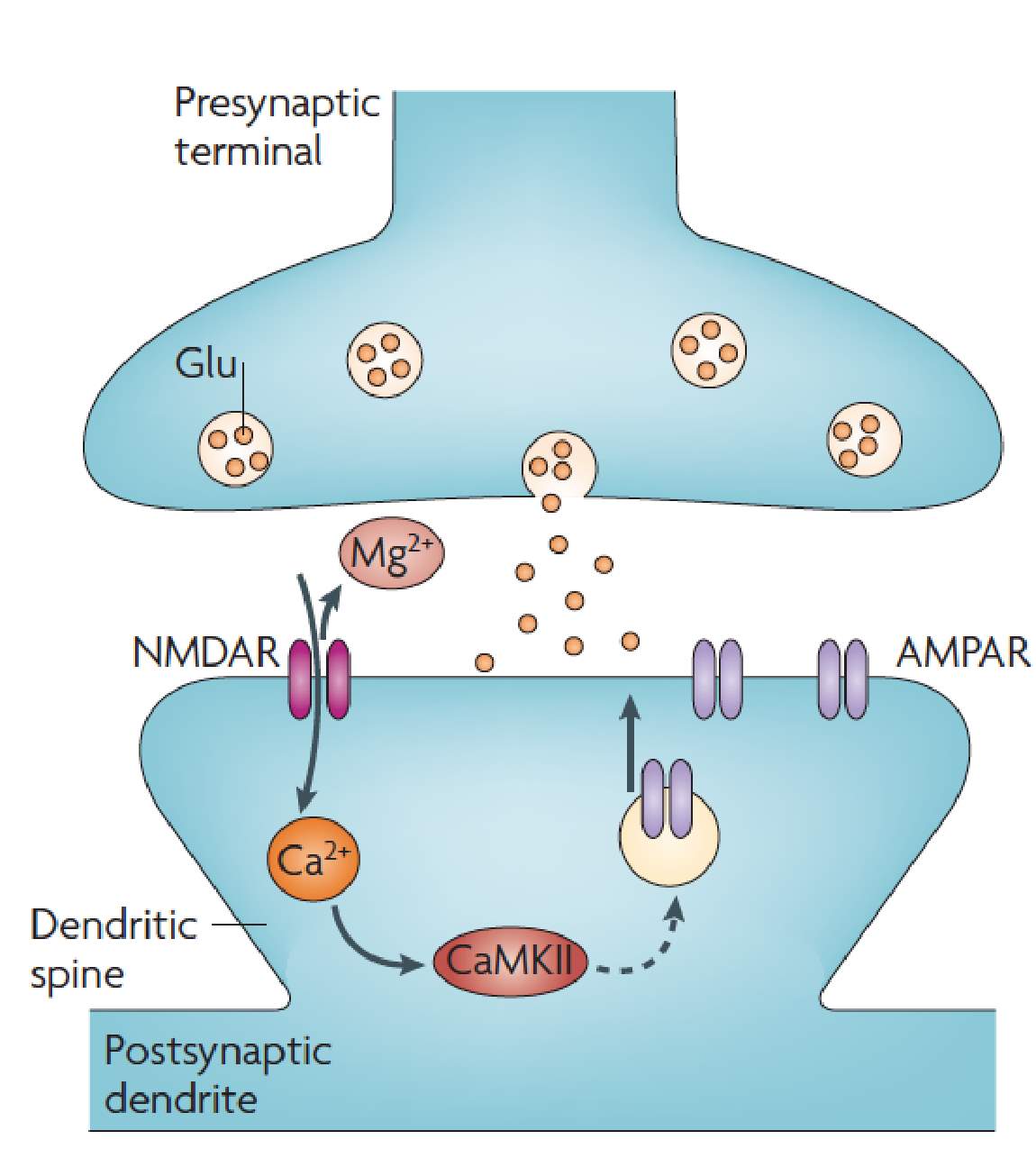
Figure 2: NMDAR dependent LTP.
Depolarisation through AMPARs relieves the Mg2+ from NMDARs. NMDARs can then initiate a number of Ca2+ dependent signaling pathways including activation of CaMKII to increase the number of AMPARs at the postsynaptic density. Image from Kauer and Malenka, 2007.
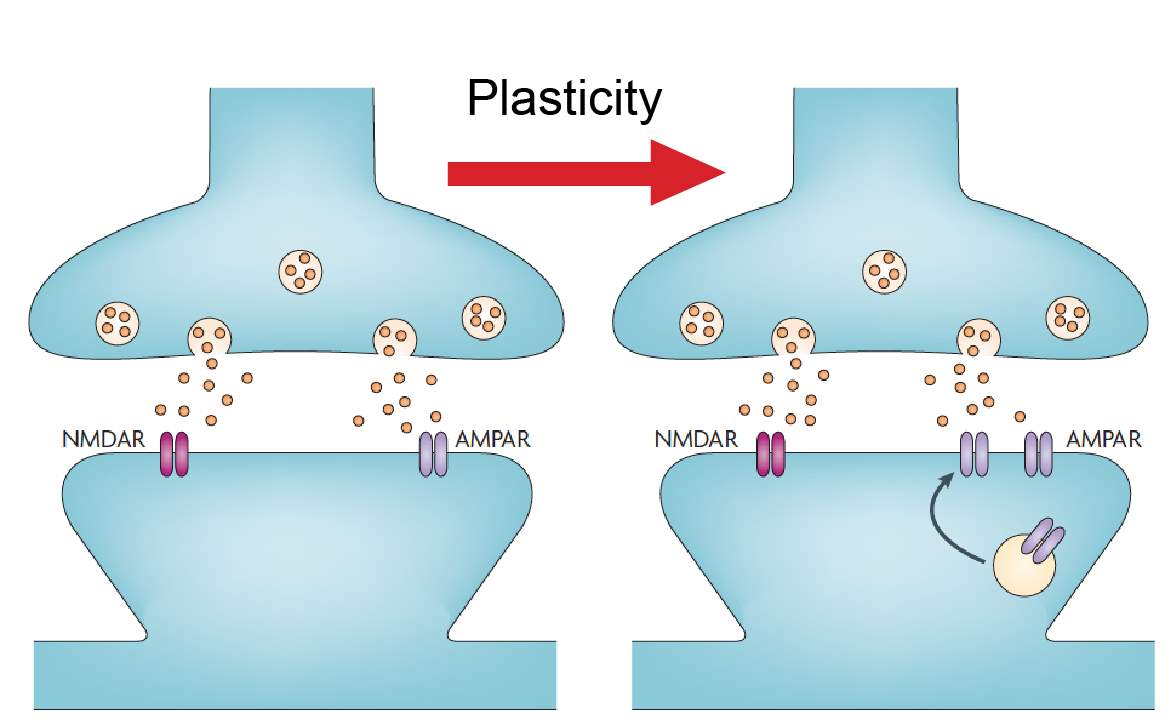
Figure 3: AMPA/NMDA ratio.
Changes in postsynaptic AMPARs can be measured using the AMPA/NMDA ratio. A synapse is shown in basal conditions and after plasticity. The number of NMDAR receptors remains unchanged after plasticity while AMPARs are increasing, increasing the AMPA/NMDA ratio. Image modified from Kauer and Malenka 2007.
1.3.2 Pain related synaptic plasticity in the Amygdala
Chronic pain conditions with ongoing peripheral injuries induce synaptic plasticity in the amygdala. Enhanced synaptic transmission of the PB-CeLC synapse has been shown following arthritis (Fu and Neugebauer, 2008, Fu et al., 2008, Han et al., 2005), spinal nerve ligation model of neuropathic pain (Ikeda et al., 2007), formalin induced inflammatory pain (Adedoyin et al., 2010), acid induced muscle pain (Cheng et al., 2011) and colitis (Han and Neugebauer, 2004). The mechanism behind this enhancement is not satisfactorily defined in arthritic conditions (Bird et al., 2005, Fu et al., 2008, Fu and Neugebauer, 2008, Han et al., 2005). However involves an increase in presynaptic glutamate release in formalin induced inflammatory pain (Adedoyin et al., 2010), an increase in postsynaptic AMPA receptor function in the spinal nerve ligation model of neuropathic pain (Ikeda et al., 2007) and an increase in number of postsynaptic AMPA receptors in acid induced muscle pain (Cheng et al., 2011). The BLA-CeLC synapse is also potentiated by an undefined mechanism during neuropathic (Ikeda et al., 2007) and arthritic pain (Ren et al., 2013, Ren and Neugebauer, 2010, Neugebauer et al., 2003). However, unlike the PB-CeLC synapse, the BLA-CeLC synapse is not potentiated in animal models of colitis (Han and Neugebauer, 2004). Synaptic plasticity at PB-CeLC synapse correlates with increased pain hypersensitivity (Fu et al., 2008, Ikeda et al., 2007, Adedoyin et al., 2010, Han and Neugebauer, 2004). The increase in AMPAR function at the PB-CeLC synapse in response to neuropathic pain correlates with allodynia (Ikeda et al., 2007). This is the same for formalin induced inflammatory pain, where the increase in presynaptic glutamate release correlates with increase in mechanical hypersensitivity and allodynia (Adedoyin et al., 2010) and arthritis which increases vocalization and hindlimb withdrawal to mechanical stimuli (Fu et al., 2008, Fu and Neugebauer, 2008, Han et al., 2005). Pharmacological inhibition of the amygdala inhibits both synaptic plasticity and pain hypersensitivity (Fu et al., 2008, Adedoyin et al., 2010), suggesting that synaptic plasticity influences the expression of pain phenotype. These data provide evidence that the amygdala is potentiated in pain models with ongoing peripheral injury. In all the above studies, the injuries are present until the animal dies. Formalin produces swelling of the paw, which is present one day later when the experiments are performed (Adedoyin et al., 2010). This is the same for the arthritis model, where inflammation and swelling of the knee takes 1-3 hours to develop, but once developed is present until the animal is euthanised 6 hours later (Fu et al., 2008, Fu and Neugebauer, 2008, Han et al., 2005). The spinal nerve ligation model of neuropathic pain results in deformity of the affected paw (Ikeda et al., 2007), which is present until the animal dies. In acid induced muscle pain, the animal is euthanised 2 hours after acid injection into the gastrocnemius muscle (Cheng et al., 2011). Thus, none of these studies address whether synapses can be potentiated when the injury has healed before the animal died. This would require a stimulus that is able to activate the pain pathways but not produce ongoing activation of peripheral nociceptors.
In summary, synaptic plasticity underlines pain hypersensitivity and the amygdala particularly the PB-CeLC synapse undergoes synaptic plasticity in pain conditions with ongoing peripheral injury. Very little is known about whether synapses such as the PB-CeLC synapse can undergo synaptic plasticity in conditions where there is no ongoing peripheral injury.
The next section will outline potential neuromodulators of this synapse and how they could influence the modulation of nociceptive processing at PB-CeLC and BLA-CeLC synapse.
1.4 Opioids
Opium extracted from poppy seeds has been used for centuries to treat a wide range of ailments including cough, diarrhea and pain (Kieffer, 1999). The active ingredient in opium, opioids are still the most effective treatment of pain but have many adverse effects (Kieffer, 1999, Inturrisi, 2002). Opioid receptors and their endogenous ligands are widely expressed in the CNS (Williams et al., 2013, Raynor et al., 1994, Dickenson, 1991), where they exert a number of effects, including activation of the endogenous descending pain pathway (Helmstetter and Fanselow, 1987, Watkins and Mayer, 1982, Helmstetter and Landeira-Fernandez, 1990). This next section will outline how opioids and their receptors exert their pharmacological actions, their use as analgesics and their expression and activity in the PB and amygdala.
1.4.1 Opioid receptors and endogenous peptides
Differential effects of opioid agonists first suggested the existence of multiple opioid receptors (Martin, 1979). It is now known that there are three opioid receptors (Kieffer, 1999, Williams et al., 2013, Raynor et al., 1994). The μ (mu) (MOR) and κ (kappa) (KOR) are found in the plasma membrane of the soma and dendrites while the δ (delta) (DOR) is often located intracellularly in the cytoplasm under control conditions (Raynor et al., 1994, Cheng et al., 1997, Cahill et al., 2001a, Arvidsson et al., 1995a, Arvidsson et al., 1995b). The endogenous opioid peptides are endorphins, enkephalins and dynorphins (Poulin et al., 2006, Dickenson, 1991).
1.4.2 Opioid Analgesia
The three opioid receptors all display antinociceptive effects when activated by their agonists (Matthes et al., 1996, Sora et al., 1997b, Tian et al., 1997, Simonin et al., 1998, Sora et al., 1997a). The MOR has the best antinociceptive properties and most commonly used opioid drugs exert their effects through this receptor (Kieffer, 1999, Pradhan et al., 2011). Deletion of MOR showed that morphine, a commonly administered opioid drug which binds to all three opioid receptors (Raynor et al., 1994, Matthes et al., 1998), only exerts its analgesic properties through MOR (Matthes et al., 1996, Sora et al., 1997b, Tian et al., 1997, Loh et al., 1998). The DOR appears to be more efficacious in chronic conditions such as neuropathic and inflammatory pain, displaying little effect in acute pain conditions (Fraser et al., 2000, Holdridge and Cahill, 2007, Mika et al., 2001, Petrillo et al., 2003, Cahill et al., 2003). This is because in many neurons DOR is mostly located intracellularly in the cytoplasm (Cahill et al., 2001a) and requires certain conditions such as inflammatory pain (Cahill et al., 2003) or chronic morphine treatment to be trafficked to the plasma membrane (Chieng and Christie, 2009, Cahill et al., 2001b). The KOR has good analgesic properties when activation is restricted to the periphery (Vanderah et al., 2008, Vanderah et al., 2004) but produces dysphoria in humans (Pfeiffer et al., 1986) and conditioned place aversion and anxiety in animal models (Land et al., 2008, McLaughlin et al., 2003, Van’t Veer and Carlezon, 2013, Mucha and Herz, 1985, Funada et al., 1993). This has restricted the use of KOR agonists in the treatment of pain and effort has been made to find KOR agonist drugs that are restricted to the periphery (Vanderah, 2010, Vanderah et al., 2008, Vanderah et al., 2004).
Opioids act by reducing both pain intensity and the emotional unpleasantness of pain (Zhang et al., 2013, Price et al., 1985, Kupers et al., 1991, LaGraize et al., 2006, Oliveras et al., 1986, Thomas et al., 1992, Gregoire et al., 2014). Interestingly, in some studies opioids only act on pain intensity at high doses (Zhang et al., 2013, Price et al., 1985, Kupers et al., 1991, LaGraize et al., 2006). Whereas, in people given low dose morphine they report reduced pain affect ratings but no change in the perceived intensity of their pain (Kupers et al., 1991, Price et al., 1985). Likewise, in rats, low dose systemic administration and intra-amygdala and intra-ACC administration of opioids reduces conditioned place aversion but has no effect on mechanical thresholds (Zhang et al., 2013, LaGraize et al., 2006). However, administration of high dosages of opioids increases mechanical thresholds to baseline levels (Zhang et al., 2013, LaGraize et al., 2006). Thus, while opioids reduce both pain intensity and affective components of pain, they have a preference for the affective component of pain.
1.4.3 Opioid receptor effectors
Opioid receptors couple to the pertussis toxin (PTX) sensitive G-protein of the Gαi and Gαo families (Connor and Christie, 1999, Williams et al., 2001). G-proteins have three subunits α, β and γ. Activation of G-protein receptors by agonists catalyze the release of GDP from Gα which is replaced with GTP. Binding of GTP leads to dissociation of Gα and Gβγ (Oldham and Hamm, 2008). The Gα and Gβγ subunits of the opioid receptor interact with cellular effectors to produce a variety of effects, the most well defined of these are inhibition of adenylyl cyclase, activation of potassium channels and inhibition of voltage-gated calcium channels (Williams et al., 2001).
1.4.3.1 Inhibition of adenylyl cyclase
Adenylyl cyclase stimulation leads to the formation of cAMP which acts within the cell to regulate a variety of cellular processes (Williams et al., 2001). Opioid inhibition of adenylyl cyclase was first seen in rat brain and cell lines (Sharma et al., 1975, Collier and Roy, 1974, Cooper et al., 1982). Opioid inhibition of adenylyl cyclase has two main effects on neurons. The first is inhibition of hyperpolarisation activated cation channels (Ih) (Williams et al., 2001). Ih is a non-selective cation channel that is activated by hyperpolarised membrane potentials and in some neurons, contributes to setting the resting membrane potential (Banks et al., 1993, Biel et al., 2009, DiFrancesco and Tortora, 1991, Ingram and Williams, 1994, McCormick and Pape, 1990). Ih is activated by cAMP (Banks et al., 1993, Biel et al., 2009, DiFrancesco and Tortora, 1991, Ingram and Williams, 1994, McCormick and Pape, 1990) and opioid inhibition of adenylyl cyclase and in turn cAMP production shifts Ih activation to more negative voltages, reducing the hyperpolarisation by Ih (Ingram and Williams, 1994, Svoboda and Lupica, 1998). This effect of opioids on Ih has not been studied in the CeLC neurons. The second effect of opioid inhibition of adenylyl cyclase is inhibition of neurotransmitter release (Shoji et al., 1999, Ingram et al., 1998, Chieng and Williams, 1998). This adenylyl cyclase dependent inhibition of neurotransmitter release occurs during morphine withdrawal in the nucleus accumbens (Chieng and Williams, 1998), PAG (Ingram et al., 1998) and ventral tegmental area (VTA) (Shoji et al., 1999). It has not been found at the PB-CeLC synapse.
1.4.3.2 Activation of potassium channels
Opioid receptors activate at least three types of potassium channels; G-protein activated inwardly rectifying potassium (GIRK) channels (Chieng and Christie, 1994, Chieng et al., 2006), voltage dependent potassium channels (Vaughan et al., 1997, Faber and Sah, 2004) and calcium-sensitive potassium channels (Twitchell and Rane, 1993). The most well-studied is opioid activation of GIRK channels. GIRKS are part of a family of inwardly rectifying potassium channels (Kir1-Kir7). The inward rectification of GIRK channels is due to occlusion of the channel by magnesium and polyamines at voltages above the potassium equilibrium potential (Wickman et al., 1994, Reuveny et al., 1994, Luscher and Slesinger, 2010). Opioids activate GIRK channels by direct binding of Gβγ subunit to GIRK channels (Reuveny et al., 1994, Huang et al., 1995, Wickman et al., 1994). Activation of GIRKs results in a voltage dependent outward flow of potassium ions, leading to hyperpolarisation of neurons (Williams et al., 2001). Opioids activate the GIRK conductance in a subpopulation of CeLC neurons (Chieng et al., 2006).
1.4.3.3 Inhibition of voltage-gated calcium channels
Voltage gated calcium channels (VGCC) are important for stimulating neurotransmitter release (Katz and Miledi, 1967a, Katz and Miledi, 1967b, Borst and Sakmann, 1996). They are found in all nerve cells and contain five classes; L-type, P/Q-type, R-type and T-type (Kandel, 2013). In neurons, it is the P/Q and N-type that mediate release of neurotransmitters (Kandel, 2013). Opioids inhibit VGCC activity by binding of Gβγ to VGCC, and as a result can reduce neurotransmitter release (Moises et al., 1994, Acosta and Lopez, 1999, Wu et al., 2004).
1.4.4. Opioid expression in the Parabrachial nucleus
The lateral parabrachial nucleus expresses all three opioid receptors albeit at varying levels (Chamberlin et al., 1999, Ding et al., 1996, Erbs et al., 2015, Arvidsson et al., 1995a, Mansour et al., 1995, Mansour et al., 1994). The lateral PB neurons also express the opioid receptor mRNAs, indicating that the neurons can synthesise the receptors (Mansour et al., 1995, Mansour et al., 1994, Le Merrer et al., 2009). The lateral PB has intense labeling for the MOR (Chamberlin et al., 1999, Ding et al., 1996, Erbs et al., 2015) and it’s mRNA (Mansour et al., 1995, Mansour et al., 1994, Le Merrer et al., 2009). The DOR and DOR mRNA expression is light but is consistently observed across studies (Arvidsson et al., 1995a, Mansour et al., 1995, Mansour et al., 1994, Erbs et al., 2015). KOR and KOR mRNA are expressed at low to moderate levels in the lateral PB (Mansour et al., 1995, Mansour et al., 1994). Therefore, the lateral parabrachial neurons that project to the CeLC contain all opioid receptors and can regulate this synapse.
1.4.5 Opioid expression in the Amygdala
The BLA and CeA (including the CeLC) both have moderate levels of MOR expression (Ding et al., 1996, Poulin et al., 2006, Le Merrer et al., 2009, Erbs et al., 2015) and it’s mRNA (Mansour et al., 1995, Mansour et al., 1994, Poulin et al., 2006, Le Merrer et al., 2009). DOR and it’s mRNA are expressed at high levels in the BLA (Mansour et al., 1995, Mansour et al., 1994, Le Merrer et al., 2009, Poulin et al., 2006) but have not been detected in the CeA (Le Merrer et al., 2009, Erbs et al., 2015, Poulin et al., 2006). The BLA and CeA both have moderate to high levels of KOR expression (Unterwald et al., 1991) and KOR mRNA (Mansour et al., 1995, Mansour et al., 1994, Le Merrer et al., 2009). Thus, both the BLA and CeA can be regulated by opioids. The BLA projections to the CeLC can potentially be regulated by all three opioid receptors, while MOR and KOR may modulate the CeA’s output.
1.4.6 Opioid activity in the PB and amygdala
There is evidence of opioid modulation of neurons in the PB and amygdala, however whether opioids modulate the PB-CeLC and BLA-CeLC synapse has not been investigated. Systemic morphine depresses pain induced neuronal responses in the lateral PB and CeA (including CeLC) (Huang et al., 1993a, Huang et al., 1993b), however this morphine effect could be indirect. Electrophysiology recordings in single neurons in the PB and amygdala have provided more direct evidence that opioids have effects in these structures (Christie and North, 1988, Finnegan et al., 2005, Zhu and Pan, 2004, Zhu and Pan, 2005, Chieng et al., 2006, Blaesse et al., 2015). In the lateral PB, despite the expression of all three opioid receptors, only MOR activation hyperpolarises lateral PB neurons (found in 97% of lateral PB neurons) (Christie and North, 1988). This suggests any potential opioid regulation of synaptic transmission at the PB-CeLC synapse is likely through MOR. Although, as mentioned earlier, the DOR is mainly located intracellularly in the cytoplasm under control conditions (Cahill et al., 2001a) and thus, may also regulate this synapse in other conditions. In the amygdala, although the BLA has high expression of DOR (Le Merrer et al., 2009, Erbs et al., 2015), it is only MOR activation that inhibits presynaptic glutamate release at the BLA-CeM synapse (Zhu and Pan, 2005). It remains to be seen whether this is also the case for BLA projections to CeLC neurons. While the direct effect of opioids on the PB-CeLC and BLA-CeLC synapse is unknown, the effect of opioids on CeA output neurons is more well-known. All three opioid receptors can hyperpolarise CeA neurons through activation of GIRK channels (Chieng et al., 2006, Zhu and Pan, 2004). However, within the CeLC subdivision of the CeA it is only the MOR that is coupled to GIRKs (Chieng et al., 2006).
Opioids are the mainstay treatment of pain. They reduce pain by inhibiting the intensity of pain and the affective component of pain. Opioid receptors are not only present in the PB, BLA and CeA; they also appear to have activity in these regions. However, it is unknown whether opioids regulate the two synapses important for pain affect, the PB-CeLC and BLA-CeLC synapse.
1.5 Calcitonin gene-related peptide (CGRP)
1.5.1 CGRP expression in the PB-CeLC synapse
CGRP is a 37-amino acid peptide that is a product of alternative splicing of the gene for calcitonin (Russell et al., 2014, Walker et al., 2010, Doods et al., 2007). CGRP peptide is so abundant in the PB terminals, that it is used as a marker for PB terminals in the CeLC (Carter et al., 2013, Chieng et al., 2006, Han et al., 2015, Haring et al., 1991, Kruger et al., 1988, Shimada et al., 1985) and CeLC neurons express CGRP receptors (van Rossum et al., 1997, Oliver et al., 1998, Han et al., 2015). CGRP terminals from the PB form asymmetric (glutamatergic) synapse with CeLC dendritic shafts and spines but also form symmetric (non-glutamatergic) synapses with CeLC soma (Dong et al., 2010, Lu et al., 2015). The presence of symmetric synapses between CGRP terminals and CeLC soma suggest that release of CGRP from these terminals could have a direct postsynaptic effect on CeLC neurons.
1.5.2 CGRP receptor signaling
The CGRP receptor is a G-protein coupled receptor (Russell et al., 2014, Walker et al., 2010, Doods et al., 2007). CGRP receptors consist of three components; calcitonin-receptor-like-receptor (CRLR), receptor activity modifying protein-1 (RAMP1) and receptor component protein (RCP) (McLatchie et al., 1998, Nikitenko et al., 2006, Evans et al., 2000, Ueda et al., 2001). RAMP1 confers ligand specificity and RCP is responsible for signal transduction (McLatchie et al., 1998, Nikitenko et al., 2006, Evans et al., 2000, Ueda et al., 2001). The CGRP receptor is closely related to other peptides in the calcitonin family, such as adrenomedullin receptor 1 and 2. The difference between the three receptors is the type of RAMP complex. Adrenomedullin receptor 1 and receptor 2 complex with RAMP2 and RAMP3 respectively (McLatchie et al., 1998, Nikitenko et al., 2006, Evans et al., 2000, Ueda et al., 2001). Varying CGRP results across studies initially led to suggestions of different CGRP receptor subtypes, however these discrepancies may be because, CGRP has affinity for all three of the receptors (Russell et al., 2014, Walker et al., 2010, Doods et al., 2007). The effect of CGRP is also complicated by a variable receptor signaling pathways. The best understood pathway is through Gαs activation of cAMP and PKA (Walker et al., 2010), however there is also evidence of coupling to other signaling pathways (Walker et al., 2010). The CGRP receptor may couple to Gαi/o G-proteinas activation of calcium currents and activation of c-Jun N-terminal kinase (JNK) by CGRP is found to be sensitive to PTX, which prevents Gαi/o G-protein activity (Wiley et al., 1992, Disa et al., 2000). The receptor may also couple to Gαq/11 G-protein; as CGRP has also been shown to activate the downstream target of Gαq/11 PLCβ in human cell lines (Drissi et al., 1998).
1.5.3 CGRP and pain
CGRP is a vasodilator and is a target for the treatment of migraines (Doods et al., 2007), however its involvement in pain is not limited to migraines. In inflammatory pain, injection of CGRP receptor antagonist into the spinal cord reduces mechanical hyperalgesia and allodynia (Neugebauer et al., 1996, Sun et al., 2004, Sun et al., 2003) and this inhibition is through a PKA and PKC dependent mechanism (Sun et al., 2004). While most studies have focused on the effect of CGRP at the periphery and spinal cord, it can also modulate pain through the PB-CeLC synapse. At this synapse, CGRP receptor antagonists CGRP8-37 and BIBN4096BS inhibit synaptic potentiation in brain slices from arthritic rats (Han et al., 2005). Microinjection of these antagonists into the CeLC inhibit arthritis induced pain related behaviours such as spinal reflexes and vocalizations (Han et al., 2005) and delivery of CGRP into the CeLC results in increased vocalization and reduction of hindpaw withdrawal threshold in animals with no prior tissue injury (Han et al., 2010). The abundance of PB CGRP terminals in the CeLC and the presence of synaptic contacts between these terminals and CeLC soma, makes CGRP an interesting prospect to study in terms of neuromodulators of the PB-CeLC synapse. However, CGRP’s involvement in pain particularly through the PB-CeLC makes knowledge of how CGRP modulates the PB-CeLC synapse even more significant.
1.6 Optogenetics
Optogenetics is the combination of light and genetics to precisely control and monitor the activities of neurons in brain slices and specific neuronal populations in freely moving animals (Yizhar et al., 2011, Nagel et al., 2005, Boyden et al., 2005). It has allowed neuroscientists to control neurons based on their location and neuronal type in a way that electrical stimulation could never allow (Yizhar et al., 2011, Nagel et al., 2005, Boyden et al., 2005).
In 1979, Francis Crick suggested in his editorial “Thinking about the brain”, that light might offer a way for neuroscientists to control specific cell types while leaving others untouched (Crick, 1979). Nearly a decade earlier in an unrelated field of research, bacteriorhodopsin, a light activated proton pump from halobacterium halobium, had already been discovered (Oesterhelt and Stoeckenius, 1971, Oesterhelt and Stoeckenius, 1973). Thus, the existence of microbial opsin genes was known, however it took decades for the concept of optogenetics as it is known today to become a reality. The discovery of bacteriorhodopsins led to the discoveries of other members of the microbial opsin gene family, including halorhodopsins, a chloride pump from arachaebacteria (Matsuno-Yagi and Mukohata, 1977) and channelrhodopsins from green algae chlamydomonas reinharditis (Nagel et al., 2002, Nagel et al., 2003). It wasn’t until 2005, 26 years after Professor Crick’s suggestion, that optogenetics became a reality (Nagel et al., 2005, Boyden et al., 2005). The next section will provide an overview of components of optogenetics and its limitations with a focus on channelrhodopsins-2.
1.6.1 Channelrhodopsin-2
Channelrhodopsin-2 (ChR2) is a non-selective cation channel that is activated by blue light (Yizhar et al., 2011, Nagel et al., 2005, Boyden et al., 2005). Expression of ChR2 allows cells to be depolarised by brief pulses of blue light (Yizhar et al., 2011, Nagel et al., 2005, Boyden et al., 2005). Light will only depolarise neurons which express ChR2, thus if the expression of ChR2 doesn’t spread to areas outside the region of interest, light pulses will only depolarise neurons in the region of interest (Yizhar et al., 2011, Boyden et al., 2005). This is an advantage over electrical stimulation where fibers from other regions may also be recruited during stimulation, making it difficult to ascertain the characteristics of discrete synapse populations. The use of promoters in optogenetics allows for even more specificity, as only the promoter cell type will express ChR2. An example of this is the use of synapsin to target only neurons and even more specifically CAMKII to largely target glutamatergic neurons (Yizhar et al., 2011). Wild-type ChR2 has been mutated to address some of the limitations of the channel (Yizhar et al., 2011, Lin, 2011). In order to improve expression levels, algal codons were replaced with mammalian codons to produce a humanized ChR2 (HChR2) (Yizhar et al., 2011). To increase the light evoked current amplitude, a gain of function mutation was introduced (H134R) (Nagel et al., 2005). ChR2(H134R) improved current amplitude however slowed the ChR2 kinetics (Yizhar et al., 2011, Lin, 2011). The ultrafast ChR2 mutations, which can response to high frequency stimulation (up to 200 Hz) were introduced to address the limitations of ChR2(H134R). Ultrafast mutations were achieved by modifying the E123T residues (Gunaydin et al., 2010). The E123T mutation produced much faster kinetics however it reduced photocurrent amplitude and light sensitivity (Gunaydin et al., 2010, Lin, 2011). To achieve a compromise between kinetics and photocurrent amplitude, a double mutation containing the E123T mutation and a mutation at the T159C residue was introduced (Berndt et al., 2011). The E123T/T159C double mutation produces depolarisations at frequencies up to 60 Hz (Berndt et al., 2011). There are now numerous ChR2 mutations available to researchers, each with its own advantages and disadvantages. The decision on which ChR2 to use depends on the experimental question and the region of interest. For example some cell types will not respond at high frequencies, thus the use of ChR2 (H134R) may suffice (Yizhar et al., 2011).
1.6.2 Inhibitory optogenetics control
Optogenetics also offer the opportunity to inhibit neurons. Inhibitory control of neurons is important, as it provides insight into the importance of activity in the targeted region (Yizhar et al., 2011). Halorhodopsins from archaebacteria (Matsuno-Yagi and Mukohata, 1977) desensitized too rapidly to be considered a viable option (Zhang et al., 2007). Another type of halorhodopsin from Natronomonas pharanos (NpHR) discovered in 1982 (Lanyi and Oesterhelt, 1982), proved to be a more viable option (Zhang et al., 2007). NpHR is a chloride pump activated by red light, making it possible to be used in conjunction with ChR2. NpHR however accumulated in the endoplasmic reticulum, reducing its effectiveness (Zhang et al., 2007). A mutation of NpHR lead to an enhanced version eNpHR 3.0, which had improved plasma membrane trafficking and light sensitivity (Zhao et al., 2008, Gradinaru et al., 2008). Archaerhodopsin (Arch and eArch 3.0), a light sensitive proton pump is also used for inhibitory control (Chow et al., 2010, Mattis et al., 2011).
1.6.3 Viral delivery
Viral vectors are widely used to express ChR2 and other opsin genes (Yizhar et al., 2011). Adeno-associated virus (AAV) and lentivirus (LV) are the most commonly used vectors (Yizhar et al., 2011). AAVs are more advantageous over LV because of their low immunogenicity and enhanced expression (Yizhar et al., 2011). AAV is a small non-enveloped single-strand DNA virus. Recombinant AAVs used in optogenetics are replication-defective because of replacement of their rep and cap genes by gene expression cassette (Castle et al., 2014, Di Pasquale et al., 2003). AAV vectors are usually recombinant rAAV2 pseudotyped with various serotype packing systems e.g. rAAV2/2 or rAAV2/5) (Yizhar et al., 2011, Aschauer et al., 2013, Blits et al., 2010). The different serotypes of AAV differ in terms of their expression level with some showing tropisms to certain cell types and even brain regions (Aschauer et al., 2013). Thus, as well as choosing the right ChR2, the type of AAV serotype must also be carefully selected.
1.6.4 Limitations of optogenetics
In 2011, optogenetics was chosen by Nature as the method of the year (Deisseroth, 2011). While its very well deserved, optogenetics does have its limitations. The first and foremost is the practicality of the technique. ChR2 has lower channel conductance than other membrane channels, thus a high expression is needed to obtain light evoked currents (Lin, 2011). Viral vectors require a significant time, usually at least 3 weeks to obtain high expression levels (Aschauer et al., 2013). This is an issue in experiments where animals must be a certain age or conducted at set time points. Another limitation is the reliability of the results obtained from optogenetics. Light evoked synaptic currents in ChR2 animals depressed more following consecutive stimulation compared to synaptic currents evoked by electrical stimulation in naïve animals (Cruikshank et al., 2010, Jackman et al., 2014, Zhang and Oertner, 2007). This depression may be due to the kinetics of the ChR2 protein, although there is also evidence it is an effect of AAV vectors (Jackman et al., 2014).
1.7 Summary and study aims
Chronic pain conditions result from plastic changes in pain pathways and can arise from conditions with ongoing peripheral injury or can occur spontaneously with no sign of a peripheral injury. This introduction provided evidence for the amygdala’s role in pain particularly the PB-CeLC and BLA-CeLC synapses. Both synapses are potentiated in pain conditions with ongoing peripheral injury, where there is an ongoing peripheral drive and thus causes ongoing activation of peripheral nociceptors. It is not known whether potentiation of synapses continues after the injury has healed, where there is no ongoing activation of peripheral nociceptors. I hypothesise that ongoing potentiation of synapses in the absence of peripheral nociceptor activation is behind the persistent pain in conditions such as back pain. This potentiation is regulated by neuromodulators. A greater understanding of how this potentiation occurs and how neuromodulators regulated pain synapses in normal and pain conditions will provide better therapeutic targets for the treatment of chronic pain. The first aim of this thesis was to investigate whether a brief nociceptive stimulus that can activate the spinal-parabrachial-amygdala pathway without damage and hence does not cause ongoing activation of the pathway, can produce synaptic plasticity at the PB-CeLC synapse.
Opioids are the most effective treatment available for pain. They act by reducing the pain intensity and the emotional unpleasantness of pain. While opioids and their receptors are expressed at the PB-CeLC and BLA-CeLC and have activity in the amygdala, it is not known whether opioids modulate the PB-CeLC and BLA-CeLC synapses. The second aim of this thesis was to determine whether opioids modulate these synapses.
Optogenetics provides an opportunity to selectively control neurons with light pulses however it may alter normal physiology at synapses. The third aim of my thesis, was to use optogenetics to selectively activate the PB-CeLC synapse, to investigate opioid modulation of this synapse and to determine whether optogenetics alters normal physiology at this synapse. Another potential modulator of pain transmission in the amygdala is CGRP. CGRP is abundantly expressed at the PB-CeLC synapse. CGRP terminals from the PB forms symmetric synapses with CeLC cell bodies and has been previously been shown to enhance synaptic transmission at this synapse. What is not known however is whether this peptide has direct effect on CeLC neurons. Thus, the final aim of my thesis was to determine whether CGRP modulates the PB-CeLC synapse and whether it can directly regulate CeLC neurons.
Chapter 2: Methods
2.1 General Methods
2.1.1 Animals
Male Sprague-Dawley rats (3-7 weeks old) were sourced from Animal Resources Centre, Perth. Rats were housed in a temperature-controlled environment with a 12-hour light/dark cycle and were provided with food and water ad libitum. The University of Sydney Animal Ethics Committee approved all experimental procedures.
2.1.2 Preparation of brain slices
Rats were anaesthetized with isoflurane, decapitated and their brains removed into ice-cold ACSF solution containing (in mM) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4.2H2O, 2.5 MgCl2, 0.5 CaCl2, 25 NaHCO3 and 11 glucose. Coronal slices (280 μm) containing the amygdala were obtained using the Leica VT 1200s vibratome. Slices were transferred to a submerged chamber containing 34°C ACSF equilibrated to pH 7.4 with 95% 02 and 5% CO2 for at least 1 hour. Preparation of brain slices were the same for chapters 3, 4 and 6.
2.2.3 Data Analysis
GraphPad Prism (Version 7, GraphPad, San Diego, CA, USA) was used for all statistical analysis.
2.2 Chapter 3 Methods
2.2.1 Nociceptive stimulus
Noxious heat was used as the nociceptive stimulus. Both hindpaws were immersed (up to the ankle) in a thermostated 44°C water bath for a 30 second period, four times with 2-minute intervals (Figure 4A). Control rats underwent the same protocol using a 33°C water bath. During the stimulus rats were anaesthetized using 3.5% isoflurane. Initial experiments to established the adequate depth of anesthesia found that rats withdrew their hindpaws at 44°C when under light anesthesia (< 3.5%).
2.2.2 Peripheral inflammation
To determine whether the nociceptive stimulus produces peripheral inflammation, I measured the paw volume displacement using a plethysmometer (Ugo Basile, VA, Italy). Hind paw volume displacement was measured before the nociceptive stimulus, 2 minutes after the nociceptive stimulus and 3 hours after the nociceptive stimulus. The same protocol was conducted with a 52°C water bath as a positive control (Bester et al., 1997).
2.2.3 Immunohistochemistry
Three hours or 1 day after nociceptive stimulation, rats were deeply anesthetized with sodium pentobarbitone (50mg/kg) and euthanized by transcardial perfusion with 3,000 IU 1-1 heparin in a 0.5% NaNO2/0.9% saline (wt/vol) solution followed by a 4% paraformaldehyde (wt/vol) solution in 0.1M phosphate buffered saline (PBS, pH 7.4). The brain and lumbar enlargement portion of the spinal cord were removed and post-fixed overnight in 4% paraformaldehyde in PBS at 4°C. Brain and spinal cord were sectioned coronally into 50 μm sections using the Leica VT 1000S vibratome (Leica Biosystems, Nussloch, Germany). Sections containing the amygdala, PB and L4/L5 regions of the spinal cord were collected in 0.1M PBS. Sections were incubated in a 10% normal goat serum (NGS)/0.5% bovine serum albumin (BSA)/0.3% Triton X-100 in PBS (wt/vol) for half an hour then washed using 0.1M PBS. This was followed by an overnight incubation of sections in rabbit antibody to c-Fos (1:100, Santa Cruz Biotechnology, SC-52) primary antibody in 2% NGS in PBS at room temperature. c-Fos primary antibody was washed off the following day with 0.1M PBS. Sections were incubated for 1 hour in guinea pig antibody to calcitonin gene related peptide (1:1000, Peninsula Laboratories, T-5027) primary antibody in 2% NGS in PBS at room temperature. Sections were washed with 0.1M PBS and then incubated in Alexa Fluor 488 goat anti-rabbit (1:1000, Molecular Probes, A-110088) and CY3 conjugated donkey anti-guinea pig (1:1000, Jackson ImmunoResearch, 706-165-148) in 2% NGS in PBS for 2 hours. Topro3 (nuclei stain) (1:500, Molecular Probes, T3605) was directly added to wells in the last half hour of incubation. Sections were washed with 0.1M PBS and mounted onto glass slides and cover slipped with Fluoromont-G (ProSciTech, Queensland, Australia). Sections were imaged with Zeiss LSM510 Meta confocal microscope (Carl Zeiss, Germany). A blinded observer counted the c-Fos immunoreactive neurons in lamina I/II of the spinal cord, external lateral region of the PB and the CeLC. The rat brain atlas by Paxinos and Watson was used to identify the relevant regions (Paxinos and Watson, 1986). Calcitonin gene-related peptide (Shimada et al., 1985, Kruger et al., 1988, Chieng et al., 2006) and Topro3 staining were used to help define the relevant regions.
2.2.4 Electrophysiology
Slices were transferred to a recording chamber and superfused continuously at 2.5ml/min with aCSF containing (in mM) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4.2H2O, 1 MgCl2, 2 CaCl2, 25 NaHCO3 and 11 D-Glucose saturated with carbogen. The temperature was maintained between 33°C-34°C using an inline heater and monitored using a thermistor. Slices were visualized using an Olympus BX51 microscope equipped with 40x water immersion objective and Dodt gradient contrast optics. Whole cell patch-clamp recordings were made from neurons in the CeLC. Patch electrodes (2-4 MΩ) were filled with internal solution containing (in mM) 140 CsCl, 5 HEPES, 10 EGTA, 2 CaCl2, 2 Mg2ATP, 0.3 NaGTP and 3 QX-314.Cl (pH 7.3, osmolarity 280-285 mOsm). Neurons were voltage-clamped using a patch clamp amplifier (Multi-clamp 700B, Axon instruments Foster city, CA). Current signals were filtered at 5kHz and sampled at 10kHz. Series resistance (≤ 12 MΩ) were compensated by 60% and continuously monitored throughout the experiment. Data was discarded if series resistance fluctuated by more than 20% during recording. Recordings were not corrected for liquid junction potentials. EPSCs were evoked via concentric bipolar stimulating electrodes (rate, 0.05 Hz; stimuli, 2-99V, 100 μs) (FHC, Bowdoin, ME, USA). Stimulus intensity was set to yield sub-threshold eEPSCs amplitudes. All eEPSCs were recorded in the presence of the GABAA receptor antagonist picrotoxin (100 μM). CeLC neurons were voltage-clamped at −70 mV or +40 mV for AMPA/NMDA ratio recordings. The AMPAR eEPSC amplitude was determined by measuring the peak amplitude of the eEPSC at −70 mV (average of at least 5 eEPSCs). The NMDAR eEPSC amplitude was determined by taking the average amplitude between 70-90ms after the stimulus at +40mV (average of at least 5 eEPSCs). AMPA/NMDA ratio was calculated by dividing the AMPAR eEPSCs amplitude by the NMDAR eEPSCs amplitude (Figure 5). To determine the voltage dependence of the AMPAR eEPSC the membrane potential was stepped from −70 mV to +40 mV (in 10 mV steps) during superfusion of the NMDAR antagonist DL-APV (100 μM) and after the addition of spermine (100 μM) to the internal solution. The rectification index is peak eEPSC+40mV divided by peak eEPSC−60mV. For comparison of AMPAR deactivation kinetics, eEPSCs were recorded at −70mV, averaged and the current decay fitted to a double exponential function. Weighted time constant was calculated using the following equation:
w = [Af/(Af+As)]f + [As/(Af+As)]s
Where Af = amplitude of the fast decay component, As = amplitude of the slow decay component, f = decay time constant of fast decay component, s = decay time constant of slow decay component.
Paired pulse ratio (PPR) of AMPAR mediated eEPSCs was obtained by evoking two consecutive stimuli of identical stimulus strength (interstimulus interval of 30 ms). PPR was calculated by dividing the second eEPSC amplitude by the amplitude of the first (eEPSC2/eEPSC1).All data were acquired and analysed using Axograph software (Molecular Devices).
2.2.5 Behavioural testing
2.2.5.1 Thermal Hyperalgesia
To measure thermal paw withdrawal latency (PWL) rats were placed in perspex enclosures (15 × 15 × 18 cm) and given 10- 15 minutes to acclimatise to the test environment. The testing was conducted using a plantar tester (Ugo Basile, Italy) according to the Hargreaves method (Hargreaves et al., 1988). Focal infrared heat was applied through the plastic bottom of the enclosure to the rear left hindpaw and the latency for the rat to respond by moving its hindpaw away from the noxious heat source was recorded. Experimenter was blinded to rat treatment group.
2.2.5.2 Mechanical Allodynia
To measure mechanical allodynia, mechanical paw withdrawal thresholds (PWTs) were determined with a series of Von Frey hairs (range 0.4–15 g). Rats were placed in elevated perspex enclosures (28 × 15 × 18 cm) with wire mesh bases and given 15–20 minutes to acclimatise to this environment. Each Von Frey hair was tested 6 times at random locations on the plantar surface of the left hindpaw. Von Frey hairs were pressed perpendicularly against the hindpaw and held for approximately two seconds. Testing began with the 2.0 g Von Frey hair. A withdrawal response was recorded if the hindpaw was sharply withdrawn, if any paw licking took place, or if the animal flinched upon removal of the Von Frey hair. If the animal responded, then the next heavier hair was tested. If the animal did not respond, then the next lighter hair was tested. Once there was a change in response, four more hairs were tested and the mechanical PWT was calculated using the up–down paradigm (Chaplan et al., 1994). If the animals did, or did not respond to any hairs, then the mechanical PWT was assigned as 0.2 g, or 15 g, respectively.
2.2.6 Drugs
Picrotoxin and spermine were purchased from Sigma-Aldrich (St Louis, MO, USA). DL-2-amino-5-phosphonopentanoic acid (DL-APV), a NMDA receptor antagonist and NBQX, a non-NMDA receptor antagonist were both purchased from Abcam (Cambridge, UK). Picrotoxin was added directly to aCSF. Spermine was added to internal solution. Distilled water was used to make a stock solution for APV and NBQX. The stock solution was diluted to working a concentration in aCSF immediately before use and applied by gravity driven superfusion.
2.2.7 Data Analysis
Data with normal distribution are expressed as mean ± s.e.m. Data with non-normal distribution are expressed as median. The interquartile range (difference between the 75th and 25th percentile) was used to quantify variability in non-normal data. Normality was tested using the Kolmogorov-Smirnov test. Mann-Whitney test (two-tailed), ANOVA or Student’s unpaired T-test (two-tailed) were used to test significance as appropriate. A result was considered statistically significant if P< 0.05.
2.3 Chapter 4 Methods
2.3.1 Electrophysiology
Slices were transferred to a recording chamber and superfused continuously at 2.5ml/min with ACSF containing (in mM) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4.2H2O, 1 MgCl2, 2 CaCl2, 25 NaHCO3 and 11 glucose. The temperature was maintained between 33°C-34°C using an inline heater and monitored using a thermistor. Slices were visualized using an Olympus BX51 microscope equipped with 40x water immersion objective. Whole cell patch-clamp recordings were made from neurons in the CeLC. Patch electrodes (2-4 MΩ) were filled with internal solution containing (in mM) 140 CsCl, 5 HEPES, 10 EGTA, 2 CaCl2, 2 Mg2ATP, 0.3 NaGTP and 3 QX-314.Cl (pH 7.3, osmolarity 280-285 mOsm).
Neurons were voltage-clamped at -70mV using a patch clamp amplifier (Multi-clamp 700B, Axon instruments Foster city, CA). Current signals were filtered at 5kHz and sampled at 10kHz. Current signals were filtered at 5kHz and sampled at 10kHz. Series resistance (≤ 12 MΩ) were compensated by 60% and continuously monitored throughout the experiment. Data was discarded if series resistance fluctuated by more than 20% during recording. Recordings were not corrected for liquid junction potentials. EPSCs were evoked by two consecutive stimuli (inter-stimulus interval of 50ms) of identical strength via concentric bipolar stimulating electrodes (rate, 0.05 Hz; stimuli, 2-99V, 100 μs) (FHC, Bowdoin, ME, USA). All eEPSCs were recorded in the presence of picrotoxin (100 μM) to block GABAA receptors. Data were acquired and analysed using Axograph software (Molecular Devices).
2.3.2 Drugs
Picrotoxin, a GABAA receptor antagonist was from Sigma-Aldrich (St Louis, MO, USA). Methionine-Enkephalin (Met-Enk), a MOR and DOR agonist was from Bachem AG (Bubendorf, Switzerland). ICI-174, 864 (ICI), a DOR receptor antagonist was from Tocris Bioscience (Bristol, UK). CTAP, a MOR antagonist was from Cayman Chemicals (Michigan, USA). U-69593 (U69), a KOR agonist and Nor-Binaltorphimine (Nor-BNI), a KOR antagonist was from Abcam (Cambridge, UK). Picrotoxin was added directly to aCSF while stock solutions were made for all other drugs. Stock solutions of drugs were made in distilled water except for U69, which was made in DMSO. Stock solutions were diluted to working concentrations in aCSF immediately before use and applied by gravity driven superfusion.
2.3.3 Data Analysis
Results are expressed as mean± sem and statistical significance was determined by Student’s one-tailed t test (paired or unpaired where appropriate). A result was considered statistically significant if P< 0.05.
2.4 Chapter 5 Methods
2.4.1 Stereotaxic surgeries
Anesthesia was induced in rats with 5% isoflurane and maintained with 2.5% isoflurane. Once deeply anesthetized, rats were placed in the stereotaxic apparatus (model 942, Kopf instruments, Tujunca, CA) and head shaved to reveal skin surface. Prior to the incision, a subcutaneous injection of caprofen (5mg/kg) (Cenvet, NSW, Australia) and 0.5% Bupivacaine (Cenvet) injected under the surface of the incision site was given. After the incision, two small holes were drilled above the lateral parabrachial nucleus (anteroposterior -8.5-9.2, mediolateral ± 2.5, dorsoventral -6.0 mm from bregma) (Paxinos and Watson, 1986). Bilateral injections were performed using glass pipettes (Drummond Scientific, Broomall, PA) filled with 300nl or 500nl of AAV5-HSYN-HChR2(H134R)-EYFP at a rate of 120nl/min or 200nl/min respectively (Nanoject 2000, Drummond Scientific) over 2.5 minutes. After the injection, the pipette was left in place for 5 minutes to allow diffusion of viral solution before being slowly withdrawn. Bone wax (Coherent Scientific) was used to seal the skull opening. After the surgery, rats were given 0.3ml of 300mg/ml solution of procaine penicillin (Benacillin, Cenvet) and 0.3ml of 100mg/ml solution of cephazolin (Hospira, Cenvet). Rats were monitored daily and postoperative procedures including weight and infection management was performed for the remainder of the experiment.
2.4.2 Preparation of brain slices
Three to twelve weeks after surgeries, rats were anaesthetized with isoflurane, decapitated and their brains removed into ice-cold aCSF containing (in mM) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4.2H2O, 2.5 MgCl2, 0.5 CaCl2, 25 NaHCO3 and 11 D-Glucose saturated with carbogen (95% O2/5% CO2). Coronal slices (280μm) containing the amygdala and parabrachial nucleus were obtained using the Leica VT 1200s vibratome. Slices were hemisected and initially incubated in an NMDG-HEPES recovery solution containing (in mM) 93 NMDG chloride, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 D-Glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgCl2, 0.5 CaCl2, pH 7.3, 300-310 mOsm/L heated at 34°C and saturated with carbogen for 10 minutes. Slices were then transferred to a submerged chamber containing aCSF equilibrated to pH 7.4 with carbogen at 34°C for a further 10 minutes and left at room temperature for at least 1 hour.
2.4.3 Electrophysiology
Slices were transferred to a recording chamber and superfused continuously at 2.5ml/min with aCSF containing (in mM) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4.2H2O, 1 MgCl2, 2 CaCl2, 25 NaHCO3 and 11 D-Glucose saturated with carbogen. The temperature was maintained between 33°C-34°C using an inline heater and monitored using a thermistor. Slices were visualized using an Olympus BX51 microscope equipped with 40x water immersion objective and Dodt gradient contrast optics and epifluorescence illumination. Whole cell patch-clamp recordings were made from neurons in the CeLC and external lateral PB. For recordings in CeLC, patch electrodes (2-4 MΩ) were filled with internal solution containing (in mM) 140 CsCl, 5 HEPES, 10 EGTA, 2 CaCl2, 2 Mg2ATP and 0.3 NaGTP (pH 7.3, osmolarity 280-285 mOsm). For recordings in PB, patch electrodes (2-4 MΩ) were filled with internal solution containing (in mM) 135 K-gluconate, 10 HEPES, 0.5 EGTA, 8 NaCl, 2 Mg2ATP and 0.3 NaGTP (pH 7.3, osmolarity 280-285 mOsm). Neurons were voltage or current clamped using a patch clamp amplifier (Multi-clamp 700B, Axon instruments Foster city, CA). Current signals were filtered at 5kHz and sampled at 10kHz. Series resistance (≤ 12 MΩ) were compensated by 60% and continuously monitored throughout the experiment. Data was discarded if series resistance fluctuated by more than 20% during recording. Recordings were not corrected for liquid junction potentials. EPSCs and action potentials were evoked in ChR2 expressing neurons and axons using a 473nm DPSS laser with a built in optic fibre (Ikecool, CA, USA). Light pulses were delivered to the slice through a 40x immersion objective lens and controlled by computer generated TTLs.
2.4.4 Immunohistochemistry
Following electrophysiology experiments, slices were fixed overnight in 4% paraformaldehyde in PBS at 4°C. The following day, sections were washed in 0.1M PBS. Sections were incubated in a 10% normal goat serum (NGS)/0.5% bovine serum albumin (BSA)/0.3% Triton X-100 in PBS (wt/vol) for an hour at room temperature then washed using 0.1M PBS. This was followed by an overnight incubation of sections in rabbit antibody to GFP (1:1000, Molecular Probes, Cat No: A6455) primary antibody in 1% NGS/1% Triton X-100 in PBS at 4°C. GFP primary antibody was washed off the following day with 0.1M PBS. Sections were then incubated for 2 hours in Alexa fluor goat anti-rabbit (1:1000, Molecular probes, Cat No: S231374) secondary antibody in 1% NGS/1% Triton X-100 in PBS at room temperature. Sections were washed with 0.1M PBS and mounted onto glass slides and cover slipped with Fluoromont-G (ProSciTech, Queensland, Australia). Sections were imaged with Zeiss LSM510 Meta confocal microscope (Carl Zeiss, Germany).
2.4.5 Drugs
Picrotoxin, a GABAA receptor antagonist was from Sigma-Aldrich (St Louis, MO, USA). Methionine-Enkephalin (Met-Enk), MOR and DOR agonist was from Bachem AG (Bubendorf, Switzerland). ICI-174, 864 (ICI), a DOR antagonist was from Tocris Bioscience (Bristol, UK). CTAP, MOR antagonist was from Cayman Chemicals (Michigan, USA). DL-2-amino-5-phosphonopentanoic acid (DL-APV), a NMDA receptor antagonist and NBQX, a non-NMDA receptor antagonists were both purchased from Abcam (Cambridge, UK). CGP 55845 (CGP), a GABAB receptor antagonist was also purchased from Abcam. Picrotoxin was added directly to aCSF while stock solutions were made for all other drugs. Stock solutions of drugs were made in distilled water except for U69, which was made in DMSO. Stock solutions were diluted to working concentrations in aCSF immediately before use and applied by gravity driven superfusion.
2.4.6 Data analysis
Results are expressed as mean ± sem. Statistical significance was determined by Student’s two-tailed t test (paired or unpaired where appropriate), two-tailed Wilcoxon matched pairs signed rank test, two-tailed Mann-Whitney test and one-way ANOVA followed by Tukey’s multiple comparison test. A result was considered statistically significant if P< 0.05.
2.5 Chapter 6 methods
2.5.1 Electrophysiology
Slices were transferred to a recording chamber and superfused continuously at 2.5ml/min with ACSF containing (in mM) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4.2H2O, 1 MgCl2, 2 CaCl2, 25 NaHCO3 and 11 glucose. The temperature was maintained between 33°C-34°C using an inline heater and monitored using a thermistor. Slices were visualized using an Olympus BX51 microscope equipped with 40x water immersion objective. Whole cell patch-clamp recordings were made from neurons in the CeLC. For synaptic transmission experiments, patch electrodes (2-4 MΩ) were filled with internal solution containing (in mM) 140 CsCl, 5 HEPES, 10 EGTA, 2 CaCl2, 2 Mg2ATP, 0.3 NaGTP and 3 QX-314.Cl (pH 7.3, osmolarity 280-285 mOsm). Neurons were voltage-clamped at -70mV using a patch clamp amplifier (Multi-clamp 700B, Axon instruments Foster city, CA). EPSCs were evoked by two consecutive stimuli (inter-stimulus interval of 50ms) of identical strength via concentric bipolar stimulating electrodes (rate, 0.05 Hz; stimuli, 2-99V, 100 μs) (FHC, Bowdoin, ME, USA). All eEPSCs were recorded in the presence of picrotoxin (100 μM) to block GABAA receptors. For current injections, GIRK and Ih experiments, patch electrodes (2-4 MΩ) were filled with internal solution containing (in mM) 135 K-gluconate, 10 HEPES, 0.5 EGTA, 8 NaCl, 2 Mg2ATP, 0.3 NaGTP (pH 7.3, osmolarity 280-285 mOsm). Neurons were voltage-clamped at -60mV in GIRK and Ih experiments. Action potentials were evoked by intracellular current injections of increasing magnitude. Current signals were filtered at 5kHz and sampled at 10kHz. Current signals were filtered at 5kHz and sampled at 10kHz. Series resistance (≤ 12 MΩ) were compensated by 60% and continuously monitored throughout the experiment. Data was discarded if series resistance fluctuated by more than 20% during recording. Recordings were not corrected for liquid junction potentials. Data were acquired and analysed using Axograph software (Molecular Devices).
2.5.2 Drugs
Picrotoxin, a GABAA receptor antagonist was from Sigma-Aldrich (St Louis, MO, USA). Calcitonin gene related peptide (CGRP) and its peptide antagonist; calcitonin gene related peptide fragment 8-37 (CGRP8-37) were also purchased from Sigma-Aldrich (St Louis, MO, USA). Methionine-Enkephalin (Met-Enk), a MOR and DOR agonist was from Bachem AG (Bubendorf, Switzerland). CTAP, a MOR antagonist and Forskolin, a cAMP activator were from Cayman Chemicals (Michigan, USA). BIBN4096BS (BIB), CGRP receptor antagonist was purchased from Tocris Bioscience (Bristol, UK). DL-2-amino-5-phosphonopentanoic acid (DL-APV), a NMDA receptor antagonist and NBQX, a non-NMDA receptor antagonists were both purchased from Abcam (Cambridge, UK). CGP 55845 (CGP), a GABAB receptor antagonist was also purchased from Abcam. Picrotoxin was added directly to aCSF while stock solutions were made for all other drugs. Stock solutions of drugs were made in distilled water except for Forskolin and BIB, which were made in DMSO. Stock solutions were diluted to working concentrations in aCSF immediately before use and applied by gravity driven superfusion.
2.5.3 Data analysis
Results are expressed as mean± sem and statistical significance was determined by student’s paired two-tailed t test. A result was considered statistically significant if P< 0.05.
Chapter 3: Central sensitization of the spino-parabrachial-amygdala pathway that outlasts a brief nociceptive stimulus.
3.1 Introduction
Acute pain provides important warnings about dangers in our environment. However, some people experience chronic pain, for instance lower back pain that outlasts this useful role and continues after the injury has healed. Chronic pain is debilitating to the person (Gureje et al., 1998) and costly to society (Atkinson, 2004). Our experience of chronic pain comprises somatosensory elements of location and intensity, negative emotional/aversive feelings (Elman and Borsook, 2016, Bushnell et al., 2013) and associative learning of ‘dangerous activities’ (Gureje et al., 1998, Vlaeyen, 2015). These learned associations between the aversive painful experience and our environment, such as a workplace, or activities, such as sports, result in the chronic pain sufferer limiting these activities (Vlaeyen, 2015).
Acute and chronic pain activates the amygdala (Bornhovd et al., 2002, Baliki et al., 2006). The amygdala contributes to the negative emotional value of the nociceptive sensory information (Cardinal et al., 2002, LeDoux, 2000) and the formation of an association between the aversive response and the environment in which it occurs (Gao et al., 2004, Pedersen et al., 2007, Ansah et al., 2010, Tanimoto et al., 2003). This association can be measured as pain-induced conditioned place aversion (Zhang et al., 2011a) and relies on the central nucleus of the amygdala (CeA) for its expression (Gao et al., 2004, Pedersen et al., 2007, Ansah et al., 2010, Tanimoto et al., 2003). This association between the aversive pain sensation and other sensory inputs, such as sound, smell and visual stimuli, is possible because the amygdala receives both nociceptive and polymodal sensory information (Moga et al., 1995, Sah et al., 2003, Vertes and Hoover, 2008, Canteras et al., 1994, McDonald and Mascagni, 1997, McDonald et al., 1996). The spinal-parabrachial-amygdala synaptic pathway sends purely nociceptive information from spinal cord via the external lateral portion of the parabrachial nucleus (PB) to the laterocapsular region of the central nucleus of the amygdala (CeLC) (Bernard et al., 1993, Bernard et al., 1992, Bester et al., 1997). The final synapse in this pathway, the PB-CeLC synapse, is required for pain-induced associative learning (Sato et al., 2015, Watabe et al., 2013, Han et al., 2015). CeLC neurons also receive highly processed polymodal sensory, including nociceptive information, from the basolateral nucleus of the amygdala (BLA) via the thalamus, insular cortex, prefrontal cortex, hippocampus and perirhinal cortex (Sah et al., 2003, Marek et al., 2013, Pape and Pare, 2010, Neugebauer, 2015). They also receive direct polymodal sensory information from the thalamus (Moga et al., 1995, Vertes and Hoover, 2008), entorhinal cortex (McDonald and Mascagni, 1997) and lateral occipital area (McDonald et al., 1996). Apart from any associations that are formed from interactions of this nociceptive and polymodal information in the CeLC, their GABAergic synaptic projections, via the substantia innominata dorsalis (Sld) (Bourgeais et al., 2001), project to brain regions important for affective responses to pain (Sah et al., 2003, Marek et al., 2013, Pape and Pare, 2010, Neugebauer, 2015, Rainville et al., 1997) suggesting that activation of the CeLC could contribute to the negative emotional experience of pain. Whilst the PB-CELC synapse does not participate in acute somatosensory responses to nociceptive stimuli (Han et al., 2015, Watabe et al., 2013), it does participate in elevated sensitivity during chronic pain states, such as hyperalgesia or allodynia (Hebert et al., 1999, Han et al., 2005, Carrasquillo and Gereau, 2007, Pedersen et al., 2007, Fu and Neugebauer, 2008, Ji et al., 2010, Ansah et al., 2010), which may rely on its projections to the brain region that largely drives endogenous analgesia, the periaqueductal grey (Haubensak et al., 2010, Bushnell et al., 2013, Veinante et al., 2013).
Chronic pain states without an ongoing injury, such as lower back pain, have been attributed to potentiation or sensitization of the neural circuits involved in pain (Basbaum et al., 2009, Ji et al., 2003, Latremoliere and Woolf, 2009, Woolf, 2011). Pain models with an ongoing injury potentiate synapses important for pain (Han and Neugebauer, 2004, Carrasquillo and Gereau, 2007, Ikeda et al., 2007, Chen et al., 2014, Cheng et al., 2011) through increases in AMPAR expression (Chen et al., 2014, Ikeda et al., 2007, Cheng et al., 2011). In fact, a range of chronic pain states, such as arthritis, formalin inflammatory pain and colitis, potentiate the PB-CeLC synapse (Fu et al., 2008, Ikeda et al., 2007, Adedoyin et al., 2010, Han and Neugebauer, 2004, Carrasquillo and Gereau, 2007). This potentiation correlates with increased pain hypersensitivity (Fu et al., 2008, Ikeda et al., 2007, Adedoyin et al., 2010, Han and Neugebauer, 2004) and inhibition of this synapse inhibits both synaptic plasticity and pain related behaviour (Fu et al., 2008, Adedoyin et al., 2010), suggesting that synaptic plasticity influences the experience of pain. The above studies use pain models, where the injury is present until the animal dies and thus can’t answer the clinically important question of whether potentiation of the PB-CeLC synapses or other synapses continues after the injury heals. The aim of this study was to determine whether a single, brief nociceptive stimulus that activates the spinal-parabrachial-amygdala pathway, without damage, and therefore had a known ‘off point’ for peripheral nociceptor activity can cause changes at the PB-CeLC synapse. This brief nociceptive stimulus should not produce ongoing activation of the pathway and thus any changes seen at the PB-CeLC synapse would be due to only the initial activation, not ongoing activation of peripheral nociceptors. The brief nociceptive stimulus increased AMPA receptors at PB-CeLC synapses for at least 3 days after the stimulus and showed similar mechanisms to long-term potentiation. It is possible this long-lasting potentiation could alter our emotional response to subsequent nociceptive stimuli or facilitate the formation of lifestyle limiting pain associations.
3.2 Aims
To define changes at the parabrachial-laterocapsular CeA synapse produced by a brief nociceptive stimulus and the time course of observed changes.
3.3 Results
3.3.1 A brief nociceptive stimulus.
To investigate synaptic plasticity that outlasts the noxious stimulus I wanted to use a brief stimulus that activates the spino-parabrachio-amygdaloid pathway but does not produce ongoing activation of this neural pathway. Briefly immersing the hindpaws of rats in 44°C water activates the spino-parabrachial pathway (Bester et al., 1997). I addressed whether this stimulus produces ongoing activity of this neural pathway by determining whether there was inflammation of the paw or ongoing c‑Fos production in the spino-parabrachio-amygdala neural pathway.
To test whether the noxious stimulus inflames the paw, I used a plethysmometer to measure the volume of water displaced by the paw before and after 44°C or 52°C heat treatment. I found that immersing the hindpaws of anaesthetized rats in 44°C water for a total of 2 minutes (Figure 4A) did not increase paw volume (Figure 4B). In fact, there was a small but significant decrease in paw volume 3 hours after the noxious heat treatment (one-way repeated measures ANOVA, Sidak post-hoc test, F(2,6) = 7.231, p = 0.025, n = 8 animals) (Figure 2B). In contrast, the 52°C heat treatment significantly increased paw volume 2 minutes (one way repeated measures ANOVA, Sidak post-hoc test, F(2,6) = 12.171, p = 0.019, n = 8 animals) and 3 hours after the heat treatment (one way repeated measures ANOVA, Sidak post-hoc test, F(2,6) = 12.171, p = 0.04, n = 8 animals) (Figure 4B). Therefore, 44°C heat treatment of the paws does not cause inflammation but higher temperatures do.
I used c-Fos, a marker for neuronal activation (Gao and Ji, 2009, Suwanprathes et al., 2003), to assess whether the nociceptive stimulus produces acute or ongoing activation of the spino-parabrachio-amygdaloid neural pathway. I counted the number of c-Fos positive neurons in the spinal cord, parabrachial nucleus and laterocapsular amygdala 3 hours and 1 day after the heat treatment. I compared rats treated with noxious heat 44°C versus control innocuous heat 33°C (Figure 4A). Noxious heat significantly increased cFos-IR in the external lateral parabrachial nucleus 3 hours after the stimulus (Figure 4C & Di) but this increase in c-Fos expression was not ongoing, as it had returned to control 1 day later (Figure 4C & Di). The noxious stimulus did not increase c-Fos expression in the spinal cord or amygdala (Figure 4Dii, iii). This is consistent with previous experiments using this noxious stimulus where at all noxious temperatures tested c-Fos protein expression was greater in the parabrachial than the spinal cord (Bester et al., 1997) . C-Fos protein levels peak 1-2 hours after a stimulus and return to basal levels 6-8 hours after the stimulus (Gao and Ji, 2009, Suwanprathes et al., 2003). Therefore, the increased c-Fos immunoreactivity observed in the parabrachial nucleus 3 hours after the nociceptive stimulus is likely in response to the activation of this neural pathway by the noxious stimulus 3 hours earlier. As c-Fos expression is not increased 24 hours later suggests that at least 6-8 hours before, this neural pathway has returned to control levels of activity. Therefore, both the lack of paw inflammation and brief elevations in c-Fos expression is consistent with the 44°C heat stimulus providing a brief noxious stimulus.
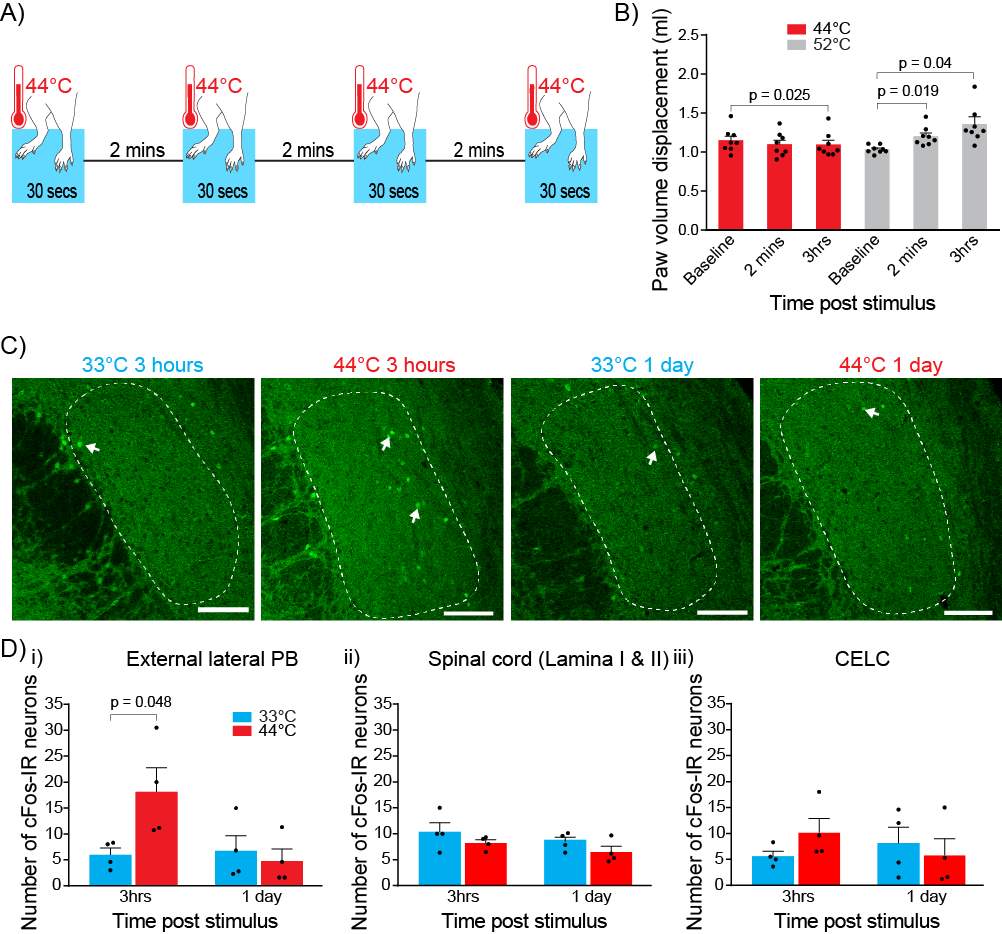
Figure 4: A brief nociceptive stimulus without inflammation or ongoing activation of the spino-parabrachio-amygdaloid pathway.
A) Nociceptive stimulus procedure. Both hindpaws were immersed in a 44°C water bath for 30 seconds. This was repeated 4x with a 2-minute inter-stimulus intervals.
B) Nociceptive stimulus does not produce peripheral inflammation measured by paw volume displacement. Paw volume was not increased 2 minutes and decreased 3 hours after 44°C nociceptive stimulus whereas paw volume was significantly increased 2 minutes and 3 hours after the positive control 52°C treatment. Statistical significance was tested with one-way repeated measures ANOVA followed by Sidak post-hoc test.
C) Confocal images of cFos-IR neurons (arrow) in the PB following heat treatment. cFos-IR neurons were counted in the external lateral portion of the PB (outlined). Rostrocaudal location of sections was as follows: 33°C 3 hours = −9.68 mm from Bregma, 44°C 3 hours = −9.8, 33°C 1 day = −9.8, 44°C 1 day = −9.68. Scale bars, 100 μm.
D) Bar charts showing number of cFos-IR neurons in the parabrachial, spinal cord and CelC following heat treatment.
i) In the external lateral Parabrachial nucleus, the nociceptive stimulus significantly increased the number of cFos-IR neurons after 3 hours but had returned to baseline levels after 1 day.
ii) Nociceptive stimulus did not alter cFos-IR in the spinal cord.
iii) Nociceptive stimulus did not alter cFos-IR in the CeLC. Statistical significance was tested with unpaired Student’s t-test. Dots show data from individual animals and bar chart shows mean ± s.e.m.
3.3.2 Nociceptive stimulus induces long-lasting synaptic plasticity specifically at PB-CeLC synapse.
I examined whether the brief nociceptive stimulus produces synaptic plasticity at the PB-CeLC synapse. A well-characterised hallmark of synaptic plasticity is changes in postsynaptic AMPARs and NMDARs (Rao and Finkbeiner, 2007). Thus, the first experiment determined the relative contribution of postsynaptic AMPARs and NMDARs (AMPA/NMDA ratio) to synaptic transmission at PB-CeLC synapse. This can be quantified using the AMPA/NMDA ratio (Figure 3 & 5). The AMPAR component was determined by measuring the peak amplitude at -70mV, as the NMDAR is blocked by magnesium at that voltage (Collingridge et al., 2004, Kauer and Malenka, 2007, Rao and Finkbeiner, 2007). This was tested using the non-NMDA receptor antagonist NBQX (10 μM). NBQX reduced EPSC amplitude at -70mV from 181.1 ± 68.03 pA in control to 14.46 ± 2.96 pA (n = 6). The NMDAR component was measured by taking the average amplitude 70-90 ms after the stimulus at +40mV. At +40mV, both AMPARs and NMDARs are active, however due to the decay kinetics of AMPARs, there is only NMDAR mediated response after 70 ms (Figure 5). Stimulating electrodes were placed dorsomedial to the central nucleus of the amygdala (CeA) to innervate PB inputs (Bernard et al., 1993, Han and Neugebauer, 2004) (Figure 6Ai). Whole-cell patch clamp recordings were then made from CeLC neurons (Figure 6Ai) and the eEPSC amplitude at −70 mV and +40 mV were recorded. The nociceptive stimulus significantly increased the AMPA/NMDA ratio at 1 day (Figure 6A & 6C). This data suggests the nociceptive stimulus increases AMPA receptors at the PB-CeLC synapses.
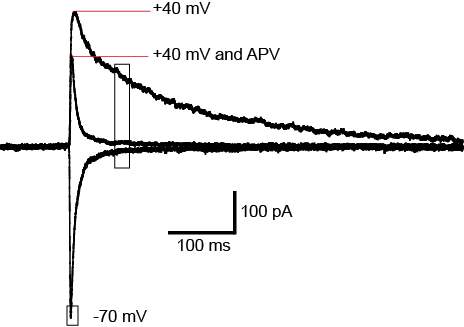
Figure 5: Measurement of AMPA/NMDA ratio.
Currents traces at -70mV, +40mV and +40mV with APV (100 μM). AMPA/NMDA was measured by taking the peak amplitude at -70mV for the AMPAR component and the average amplitude 70-90ms after the stimulus at +40mV for the NMDAR component (black box). Decay kinetics for AMPAR is shown by blocking the NMDAR receptor component with APV. Note how there is no AMPA current after 70 ms.
To determine whether the nociceptive stimulus specifically changes synapses in the PB-CeLC, I tested whether the stimulus also changes a mixed synaptic input. These mixed inputs include polymodal sensory information from the thalamus (Moga et al., 1995, Vertes and Hoover, 2008), hypothalamus (Canteras et al., 1994), entorhinal cortex (McDonald and Mascagni, 1997) and lateral occipital area (McDonald et al., 1996). They also include inputs from areas delivering affective information such as prefrontal cortex, insular cortex and anterior cingulate cortex (McDonald et al., 1996). Stimulating electrodes were placed dorsal to the CeA to stimulate this mixed input (Moga et al., 1995, Vertes and Hoover, 2008, McDonald et al., 1996, McDonald and Mascagni, 1997, Canteras et al., 1994) (Figure 6Bi). The nociceptive stimulus did not alter the AMPA/NMDA ratio at this mixed input-CeLC synapse at 1 day (Figure 6B & C). This indicates that the nociceptive stimulus does not induce synaptic potentiation at all synapses onto CeLC neurons.
The noxious stimulus only increased the AMPA/NMDA ratio in a sub-population of CeLC neurons (Fig 6C). The variability in the response may result from whether individual CeLC neurons receive noxious information in response to the stimulus as only about 40% of CeLC neurons are excited by cutaneous inputs from the PB (Bernard et al., 1992, Bernard et al., 1990). I also found that the plasticity at the PB-CeLC synapse was bilateral (44°C 1 day right hemisphere: 4.69 ± 0.98, n = 8 cells vs. 44°C 1 day left hemisphere: 6.51 ± 1.49, n = 23 cells, p = 0.49, two-tailed Student’s unpaired t-test) and was not influenced by rostrocaudal location of the neuron or the time elapsed following dissection (up to 7 hours).
As the nociceptive stimulus is brief and does not produce ongoing activation or inflammation we can track how long this pain-induced change in synaptic glutamate receptors persists after the stimulus. We found that 3 hours after the nociceptive stimulus the AMPA/NMDA ratio was unchanged from control (Figure 6D). The biggest increase in AMPA/NMDA ratio occurs 1 day and 3 days after the nociceptive stimulus (Figure 6D). By 7 days, the AMPA/NMDA ratio had returned to control levels (Figure 6D). These data show that a brief nociceptive stimulus, that does not produce ongoing peripheral activation, can induce long lasting synaptic changes at a central synapse important for pain.
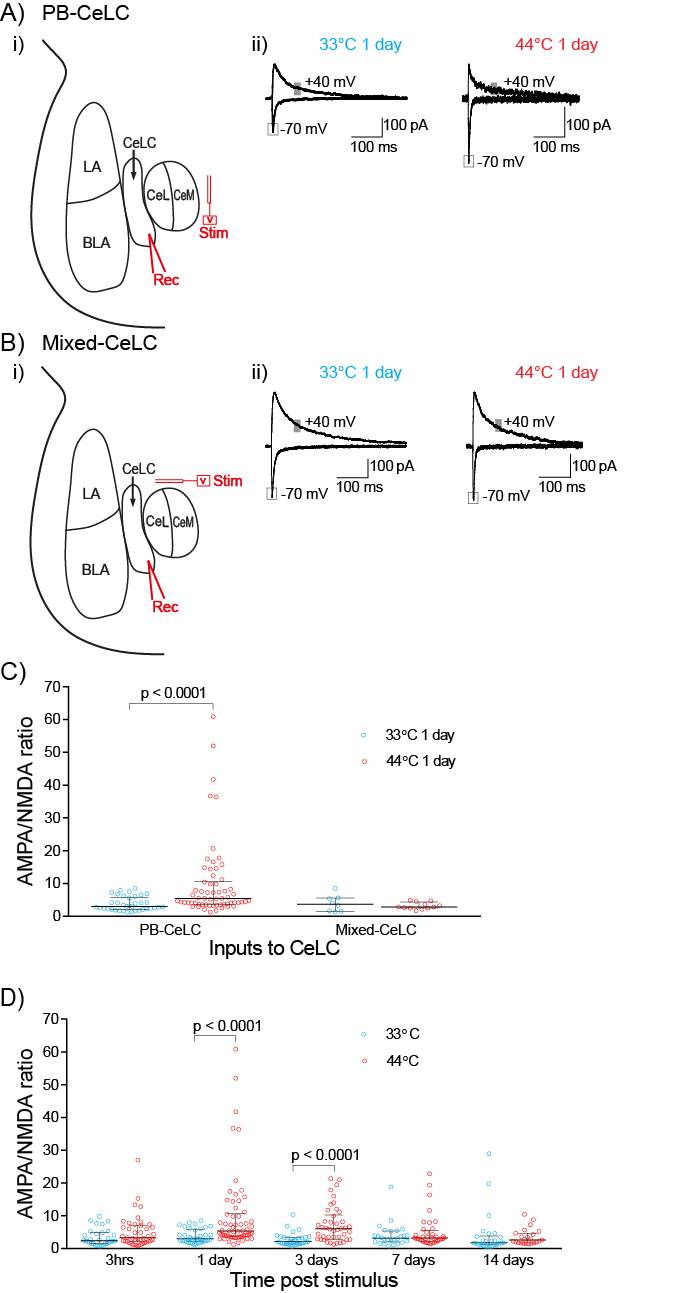
Figure 6: A brief nociceptive stimulus induces long lasting synaptic plasticity specifically at PB-CeLC synapse.
A) Nociceptive stimulus potentiates the PB-CeLC synapse.
(i) Schematic diagram of stimulation and recording site. Stimulating electrodes were placed dorsomedial to the CeA to stimulate PB fibers. The response of the CeLC neurons to this stimulation was recorded.
(ii) Example traces of eEPSCs from control and nociceptive treated rats 1 day after treatment. The amplitude of the AMPAR component of the eEPSC was measured at the peak current recorded at −70mV and the amplitude of the NMDAR component of the eEPSC was measured as the average amplitude 70-90ms after stimulation recorded at +40mV (grey box).
B) Nociceptive stimulus does not potentiate the mixed synaptic input.
(i) Schematic diagram of stimulation and recording site. Stimulating electrodes were placed dorsal to CeA to stimulate fibers coming from but not limited to the cortex, hypothalamus, thalamus and PB. The response of the CeLC neurons to this stimulation was recorded.
(ii) Example traces of eEPSCs from control and nociceptive treated rats 1 day after treatment. The amplitude of AMPAR and NMDAR eEPSCs was measured as above.
C) Scatter plots of AMPA/NMDA ratio at the PB-CeLC and Mixed-CeLC synapses for individual neurons. Nociceptive stimulus increased the AMPA/NMDA ratio at PB-CeLC synapse but not at the mixed input-CeLC synapse.
D) Nociceptive stimulus causes long-lasting changes at PB-CeLC synapse. Scatter plot showing AMPA/NMDA over the 2 weeks following the nociceptive stimulus. The nociceptive stimulus increased the AMPA/NMDA ratio for at least 3 days. Statistical significance was tested with two-tailed Mann Whitney test. Each circle shows the data from an individual neuron and graph also shows the median ± the interquartile range.
3.3.3 Nociceptive stimulus produces a transient change in AMPAR subunit composition at PB-CeLC synapse.
The increase in AMPA/NMDA ratio at the PB-CeLC synapse may be due to either an increase in postsynaptic AMPARs or a decrease in NMDARs. AMPARs are heterotetramers composed of GluA1-4 subunits and the composition of synaptic AMPA receptors changes during some forms of synaptic plasticity (Chater and Goda, 2014). In some forms of in vitro long-term potentiation (LTP), a model for learning and memory (Chater and Goda, 2014) synaptic AMPARs undergo a change in subunit from GluA2-containing to GluA2-lacking (Plant et al., 2006, Guire et al., 2008, Morita et al., 2014). Unlike GluA2-containing AMPARs, GluA2-lacking AMPARs are Ca2+ permeable (Chater and Goda, 2014), have faster decay kinetics (Chater and Goda, 2014) and display inwardly rectifying current-voltage (I/V) relationship due to blockade by intracellular polyamines (Chater and Goda, 2014). To elucidate whether the increase in AMPA/NMDA ratio was associated with a change in AMPAR subunits, I/V relationships of AMPAR eEPSCs at 1 day and 3 day time points was examined. To isolate the AMPAR synaptic response, experiments were conducted in the presence of the GABAA antagonist pictrotoxin (100 μM) and NMDA receptor antagonist DL-APV (100 μM). Spermine (100 μM) was added to the intracellular solution to compensate for possible dialysis of intracellular polyamines during whole cell recording. The nociceptive stimulus significantly increased the inward rectification of AMPAR mediated eEPSCs at 1 day (Figure 7A). This is consistent with the nociceptive stimulus increasing incorporation of GluA2-lacking AMPARs at the PB-CeLC synapse. Interestingly, although there was a significant increase in AMPA/NMDA ratio in the nociceptive group at 3 days, the rectification index of the nociceptive stimulus group at 3 days did not differ from control, suggesting that 3 days after the stimulus the AMPAR subunit composition had reverted to GluA2-containing AMPARs (Figure 7A) even though the increase in AMPA/NMDA receptor persists.
The nociceptive stimulus quickened the AMPAR decay at 1 day (Figure 7B) but the decay did not differ from the control group by 3 days post stimulus (Figure 7B). Both the increase in inward rectification and quicker decay of the AMPAR synaptic response after the nociceptive stimulus suggests that the stimulus triggers the transient insertion of GluA2-lacking AMPAR at the PB-CeLC synapse for 1 day. After this, although the AMPA/NMDA receptor ratio is elevated the AMPAR subunit composition of the PB-CeLC synapse returns to it baseline composition similar to LTP (Plant et al., 2006, Guire et al., 2008, Morita et al., 2014).
I next examined whether the nociceptive stimulus causes changes in pre-synaptic glutamate release at PB-CeLC synapse. The nociceptive stimulus did not change the paired pulse ratio (PPR) 1 day after the stimulus (nociceptive stimulus group: 1.01 ± 0.054, n = 49 cells vs. control group: 1.13 ± 0.12, n = 36 cells, p = 0.36, two-tailed unpaired Student’s t-test), suggesting that the nociceptive stimulus does not alter glutamate release (Figure 7C).
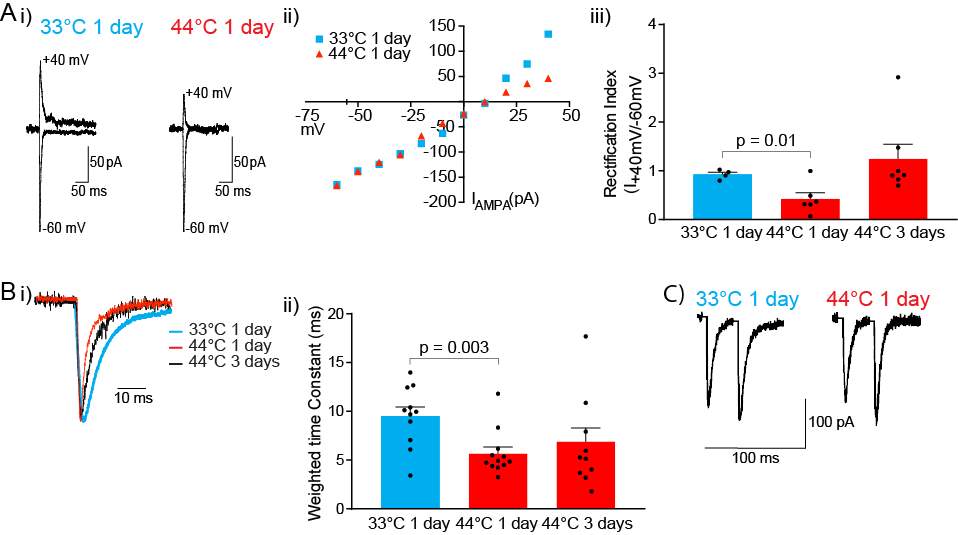
Figure 7: A brief nociceptive stimulus produces a transient change in AMPAR subunit composition at PB-CeLC synapse.
A) Nociceptive stimulus increases inward rectification of AMPAR eEPSCs at 1 day.
(i) Example traces of AMPAR eEPSCs recorded at −60mV and +40mV after control or nociceptive treatment. Recordings were made in the presence of APV (100 μM) and spermine (100 μM) was included in the internal solution.
(ii) Current-voltage plot showing greater inward rectification of AMPAR eEPSCs 1 day after the nociceptive stimulus.
(iii) Bar chart of rectification index (I+40mV/I−60mV) showing significantly lower rectification index in nociceptive stimulus group at 1 day. Rectification index returns to control levels at 3 days.
B) AMPAR mediated eEPSCs have faster decay kinetics in nociceptive group at 1 day and return to control levels after 3 days.
(i) Normalised example traces of AMPAR eEPSCs after control and nociceptive treatments.
(ii) Bar chart of weighted time constants showing that the nociceptive stimulus speeds decay of the AMPAR synaptic at 1 day.
C) PPR was not changed by the nociceptive stimulus. Example traces of two consecutive EPSCs of identical intensity (interstimulus interval of 30 ms) showing that the PPR was not changed by the nociceptive stimulus. PPR was calculated by dividing the second eEPSC amplitude by the amplitude of the first. Statistical significance was tested using two-tailed unpaired Student’s t-test. Dots show data from individual neurons and bar chart shows mean ± s.e.m.
3.3.4 PB-CeLC synapse undergoes metaplastic-like changes
Metaplasticity denotes a higher order form of synaptic plasticity, where a “primer” synaptic activity at a particular point in time alters or changes the ability of neurons or synapses to generate subsequent plasticity (Abraham, 2008). It is possible that that the synaptic plasticity produced by the nociceptive stimulus at 1 day could influence the nature of subsequent plasticity. In particular, I wondered whether if I delivered a second nociceptive stimulus when there is more calcium permeable GluA2-lacking AMPAR at the synapse, if I would observe longer or greater synaptic plasticity than after a single stimulus. To test this, I compared the plasticity produced by two consecutive nociceptive stimuli (44°C) on day 1 and on day 2 versus a nociceptive stimulus of 44°C on day 1 and 33°C on day 2. I examined the AMPA/NMDA ratio 7 days after the last nociceptive stimulus (either day 7 or day 8) when the change in AMPA/NMDA ratio after one stimulus has returned to baseline levels (Figure 8A). Seven days after the second nociceptive stimuli the AMPA/NMDA ratio was significantly higher than 7 days after one nociceptive and one control stimuli (Figure 8). This suggests that when a second nociceptive stimulus is delivered during a time of prior synaptic potentiation this lengthens the period of the synaptic plasticity.
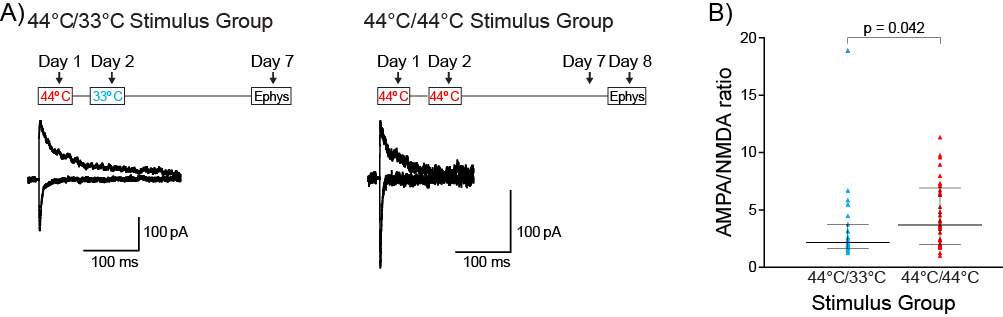
Figure 8: PB-CeLC synapse undergoes metaplastic-like changes following synaptic plasticity.
A) Nociceptive stimulus (above) and example traces of eEPSCs (below) in control two stimuli group (44°C/33°C) and nociceptive two stimuli group (44°C/44°C). The amplitude of the AMPAR and NMDAR eEPSC was measured as in figure 2.
B) Scatter plot of the AMPA/NMDA ratio showing the increase in AMPA/NMDA ratio in the nociceptive two stimuli group. Statistical significance was tested using two-tailed Mann-Whitney. Dots show data from individual neurons and graph also shows the median ± the interquartile range.
3.3.5 Nociceptive stimulus produces mechanical but not thermal hyperalgesia.
The PB-CeLC synapse contributes to development of hyperalgesia in numerous pain states (Hebert et al., 1999, Han et al., 2005, Carrasquillo and Gereau, 2007, Pedersen et al., 2007, Fu and Neugebauer, 2008, Ji et al., 2010, Ansah et al., 2010). Therefore, it was examined whether the single nociceptive stimulus induces mechanical and thermal hyperalgesia. The nociceptive stimulus significantly reduced the mechanical threshold at 1 day (Figure 9A). At all other time points the mechanical threshold was unchanged by the nociceptive stimulus (Figure 9A).The thermal threshold was unchanged at all time points (Figure 9B). Given that the nociceptive stimulus itself is a noxious thermal stimulus it may seem surprising that it produced mechanical rather than thermal hyperalgesia. However, it is consistent with secondary hyperalgesia (hyperalgesia outside of the site of injury) that is produced through central sensitization only resulting in mechanical hyperalgesia (Raja et al., 1984, Coderre et al., 1993, Dahl et al., 1993, Woolf, 2011).
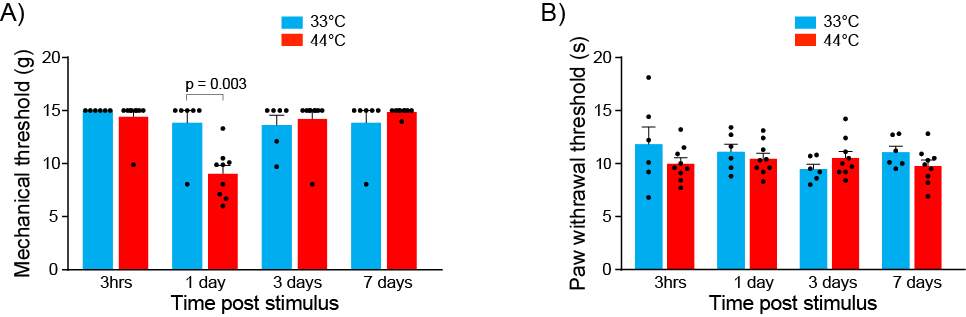
Figure 9: Nociceptive stimulus produces mechanical but not thermal hyperalgesia.
A) Bar chart of mechanical threshold showing that the single nociceptive stimulus (44°C) causes a reduction in mechanical threshold at 1 day.
B) Bar chart of paw withdrawal latency following Hargreaves test showing that the single nociceptive stimulus (44°C) does not produce thermal hyperalgesia at any time point. Statistical significance was tested using two-tailed unpaired Student’s t-test. Dots show data from individual neurons and bar chart shows mean ± s.e.m.
3.4 Discussion
In this study, I used a brief nociceptive stimulus that does not produce ongoing activation of the spino-parabrachio-amygdaloid pathway to study synaptic plasticity that persists beyond the stimulus. I found that a 2-minute nociceptive stimulus potentiates the PB-CeLC synapse. Our precise control of the nociceptive timing allowed us to determine that the increase in AMPAR response at PB-CELC synapse developed over one day and lasted for at least 3 days. Additionally, the AMPAR potentiation was biphasic with incorporation of more GluA2-lacking AMPARs at one day, which were subsequently replaced by GluA2-containing AMPARs at 3 days. I found that during the period of high GluA2-lacking AMPAR activity, an additional nociceptive stimulus produced prolonged synaptic plasticity. I also found that rats experienced mechanical hyperalgesia following the nociceptive stimulus.
It is known that a two-phase noxious stimulation of deep tissue (Cheng et al., 2011) or prolonged noxious stimulation of joints over several hours (Neugebauer and Li, 2003, Neugebauer et al., 2003, Han et al., 2005) strengthens the PB-CELC synapse for 2-6 hours. The mechanism of this potentiation is not well defined but may be due to increase in postsynaptic AMPARs (Cheng et al., 2011). The findings from the present study have allowed us to define how the glutamate receptors change over days in response to simple nociceptive activation. In vitro long-term potentiation mechanisms are thought to model synaptic modifications that underlie learning (Chater and Goda, 2014). The synaptic changes produced by the nociceptive stimulation are reminiscent of some forms of LTP (Plant et al., 2006, Guire et al., 2008, Morita et al., 2014) but stretched over a much longer time frame. Whilst in vitro high frequency stimulation increases GluA2-lacking AMPARs over minutes these receptors only remain at the synapse for 20-30 minutes and the potentiation is then maintained by subsequent replacement with GluA2-containing AMPARs (Plant et al., 2006, Guire et al., 2008, Morita et al., 2014). The nociceptive stimulus increases the AMPA/NMDA ratio during the first day post-stimulus and like the early phase of LTP the potentiation of the AMPA response is associated with greater insertion of GluA2-lacking AMPARs. By 3 days the synapses are still potentiated but the GluA2-lacking AMPARs seen at one day are replaced by GluA2-containing AMPARs by 3 days.
The role of the PB-CeLC neural pathway in acute behavioural responses to nociceptive stimuli, such as mechanical and thermal thresholds (Han et al., 2015, Sato et al., 2015) is limited but the CeA is important for the development of hyperalgesia in various pain states (Hebert et al., 1999, Han et al., 2005, Carrasquillo and Gereau, 2007, Pedersen et al., 2007, Fu and Neugebauer, 2008, Ji et al., 2010, Ansah et al., 2010). This could be through CeLC projections to the CeM, (Jolkkonen and Pitkanen, 1998, Ciocchi et al., 2010, Haubensak et al., 2010) which in turn projects to the periaqueductal gray (PAG) (Haubensak et al., 2010). The PAG controls the descending analgesic pathway (Bushnell et al., 2013, Veinante et al., 2013) and thus PB-CeLC synaptic plasticity could also mediate/alter the descending modulation of pain. Therefore, the hyperalgesia we observed 1 day after the nociceptive stimulus may result from differential activation of these pain modulatory pathways when the PB-CeLC potentiation is in place.
CeA (including CeLC) neurons have large bilateral receptive fields and display a sigmoid like stimulus-response curve to noxious stimuli (Bernard et al., 1992, Neugebauer and Li, 2002). They initially produce a graded response to increased stimulus intensity, which reaches a plateau, where increases in stimulus intensity do not produce subsequent increase in response. Whilst these characteristics would not allow appropriate sensory-discrimination of pain they are consistent with the contribution of the PB-CeLC neural pathway to pain-induced negative affect (Han et al., 2015), associative learning (Han et al., 2015; Sato et al., 2015) or regulation of pain hypersensitivity (Hebert et al., 1999, Han et al., 2005, Carrasquillo and Gereau, 2007, Pedersen et al., 2007, Fu and Neugebauer, 2008, Ji et al., 2010, Ansah et al., 2010). During the three days when there are more AMPA receptors at the PB‑CELC synapses a given nociceptive stimulus would produce unchanged release of glutamate from PB terminals in the CeLC but stronger post-synaptic depolarisation of CeLC. Thus, you would expect more neurons to reach threshold to respond to the nociceptive stimulus and ultimately greater pain-associated negative affect and facilitation of pain-induced associative learning. In addition, during the first phase after the stimulus, the increased incorporation of calcium permeable GluA2-lacking AMPAR could act as an additional source for activity dependent calcium entry and thus facilitate subsequent synaptic plasticity (Chater and Goda, 2014, Guire et al., 2008, Plant et al., 2006, Morita et al., 2014). Consistent with this I found that a second nociceptive stimulus delivered when there was high GluA2-lacking AMPAR incorporation produced longer lasting synaptic plasticity that could result from greater calcium entry through these receptors. If this is the case the you would expect that the increased negative affect would therefore persist for 3 days, like the synaptic strengthening, but any associative learning that occurred because of the stimulus-induced plasticity would persist even after this synapse was no longer potentiated. The associative learning connecting a nociceptive/noxious stimulus with the environment relies in part on activity of the PB-CeLC neural pathway (Han et al., 2015, Sato et al., 2015) and the CeA (Gao et al., 2004, Pedersen et al., 2007, Ansah et al., 2010, Tanimoto et al., 2003). The relevant plasticity is not yet defined but could be PB-CeLC assisted potentiation of other polymodal sensory inputs (Moga et al., 1995, Sah et al., 2003, Vertes and Hoover, 2008, Canteras et al., 1994, McDonald and Mascagni, 1997, McDonald et al., 1996). I found that the nociceptive stimulus did not change the AMPA/NMDA ratio for mixed inputs entering dorsal to the CeA, which contains some of the polymodal inputs to the CeLC (Moga et al., 1995, Vertes and Hoover, 2008, McDonald et al., 1996, McDonald and Mascagni, 1997, Canteras et al., 1994). This may be because the relevant synaptic plasticity for associative learning is not a change in AMPA/NMDA ratio, occurs at other synapses or did not happen in these experiments as animals had minimal sensory input during the nociceptive stimulus due to anaesthesia.
The chronic pain experience combines somatosensory experience, negative affective experience (Elman and Borsook, 2016, Bushnell et al., 2013) with fear avoidance of activities associated with pain (Gureje et al., 1998, Vlaeyen, 2015). Whilst associative learning in response to acute pain is often helpful, such as avoiding fire after a burn, in chronic pain states, the pain-associated learning is a major cause of pain related disability (Vlaeyen, 2015). In chronic pain, sufferers fear avoidance behaviours limit many activities where they have experienced pain in the past, including avoiding work and sport (Vlaeyen, 2015). The synaptic potentiation described here was in response to a brief stimulus. The potentiation lasts several days and could boost the negative affective aspect of pain during this time but the associative learning that could be facilitated by this plasticity could be much longer lasting. With repeated stimuli, the plasticity I described lasted for longer and it is possible that when chronic pain develops in response to an injury these biphasic changes in AMPARs are the initiating events.
Chapter 4: Opioids differentially modulate two synapses important for pain signaling in the amygdala
4.1 Introduction
Parabrachial inputs to the CeLC are important for the association of a painful stimulus with the environment in which it occurs (Sato et al., 2015, Watabe et al., 2013, Han et al., 2015). This association is important in the affective component of pain as it leads sufferers to limit activities they associate with pain, such as work or sport (Gureje et al., 1998, Vlaeyen, 2015). In addition to associating pain with the environment, the CeLC could also contribute to the negative emotional experience of pain through its projections via the cholinergic substantia innominata dorsalis (Sld) (Bourgeais et al., 2001), to brain regions important for affective responses to pain (Sah et al., 2003, Marek et al., 2013, Pape and Pare, 2010, Neugebauer, 2015, Rainville et al., 1997).
As mentioned previously, this synapse is potentiated in pain conditions with ongoing peripheral injury (Fu et al., 2008, Ikeda et al., 2007, Adedoyin et al., 2010, Han and Neugebauer, 2004, Carrasquillo and Gereau, 2007) and as shown in the previous chapter, is also potentiated by a brief nociceptive stimulus with no ongoing injury. Potentiation of this synapse could mean an increase in the affective responses to pain and greater associative learning of these affective responses with the environment in which they occur. Inhibition of this synapse would thus dampen this potentiation and facilitate a mechanism for reducing the affective component of pain. However, to abolish the associative learning that occurs in the affective component of pain, other pathways that deliver sensory information about the environment to CeLC neurons would also need to be inhibited. One of these pathways is the BLA projections to CeLC neurons. The BLA sends polymodal sensory information including nociceptive information to the CeLC (Sah et al., 2003, Marek et al., 2013, Pape and Pare, 2010, Neugebauer, 2015) and is also involved in pain modulation. BLA neurons respond more strongly to noxious than innocuous stimuli (Ji et al., 2010) and increase in responsiveness to innocuous stimulation after arthritis (Ji et al., 2010). Furthermore, pharmacologic inactivation of the BLA attenuates pain induced affective behaviours such as vocalization (Ji et al., 2010), suggesting that the BLA is also involved in the affective component of pain. Lesions of the BLA attenuates pain induced CPA (Tanimoto et al., 2003) and because CPA requires association of the environment with the pain stimulus, it also provides evidence that the BLA is in important for associative learning during pain. Like the PB-CeLC synapse, the BLA-CeLC synapse is also potentiated in pain conditions with ongoing peripheral injury such as neuropathic (Ikeda et al., 2007) and arthritic pain (Ren et al., 2013, Ren and Neugebauer, 2010, Neugebauer et al., 2003). This potentiation would again enhance the affective component of pain and as one of the pathways delivering sensory information to the CeLC, it would also enhance associative learning. Thus, inhibition of both the PB-CeLC and BLA-CeLC synapse would be important in reducing associative learning and the affective component of pain. In fact, modulation of both synapses may be part of the mechanism of action medications used in the clinical treatment of pain such as opioids.
Opioid modulation of the two synapses is a possibility because they act by reducing pain intensity and the affective component of pain (Zhang et al., 2013, Price et al., 1985, Kupers et al., 1991, LaGraize et al., 2006, Oliveras et al., 1986, Thomas et al., 1992, Gregoire et al., 2014) , aligning with the roles of the PB-CeLC and BLA-CeLC in pain. The three opioid receptors are also expressed in the PB and BLA (Unterwald et al., 1991, Mansour et al., 1995, Mansour et al., 1994, Arvidsson et al., 1995a, Ding et al., 1996, Chamberlin et al., 1999, Poulin et al., 2006, Le Merrer et al., 2009, Erbs et al., 2015). The PB has high expression of MOR (Chamberlin et al., 1999, Ding et al., 1996, Erbs et al., 2015), moderate levels of KOR (Mansour et al., 1995, Mansour et al., 1994) and light labelling of DOR (Arvidsson et al., 1995a, Mansour et al., 1995, Mansour et al., 1994, Erbs et al., 2015). While the BLA has intense labelling of DOR (Le Merrer et al., 2009, Erbs et al., 2015), moderate KOR (Unterwald et al., 1991) and low levels of MOR (Ding et al., 1996, Poulin et al., 2006, Le Merrer et al., 2009, Erbs et al., 2015). Opioid modulation of the PB-CeLC and BLA-CeLC is also made more likely by evidence of opioid activity in the two regions. As mentioned in the introduction of this thesis, only the MOR regulates activity in the lateral PB despite the expression of other opioid receptors (Christie and North, 1988) and in the BLA, despite the high expression of DOR, only the MOR regulates the BLA projections to the CeM (Zhu and Pan, 2005).
The PB-CeLC and BLA-CeLC synapses are both important in the affective component of pain and the associative learning involved in the affective component of pain. Opioids as the mainstay treatment of chronic pain reduce the affective component of pain. Due to the presence of opioid receptors and evidence of opioid activity in the PB and BLA, it is likely that opioids produce their antinociceptive effects by regulating the PB-CeLC and BLA-CeLC synapse. Hoverer, it is unknow if opioids directly regulate the two synapses in normal physiology. Thus, the aim of the present study was to determine the effects of opioids at the PB-CeLC and BLA-CeLC synapse. I found that opioids modulate the PB-CeLC and BLA-CeLC synapses through a pre-synaptic mechanism. Opioid modulation is via the MOR and DOR at the PB-CeLC synapse and via MOR and KOR at the BLA-CeLC synapse.
4.2 Aims
- To determine opioid regulation of the PB-CeLC synapse.
- To determine opioid regulation of the BLA-CeLC synapse.
4.3 Results
4.3.1 Met-Enk inhibits the PB-CeLC synapse through a presynaptic mechanism.
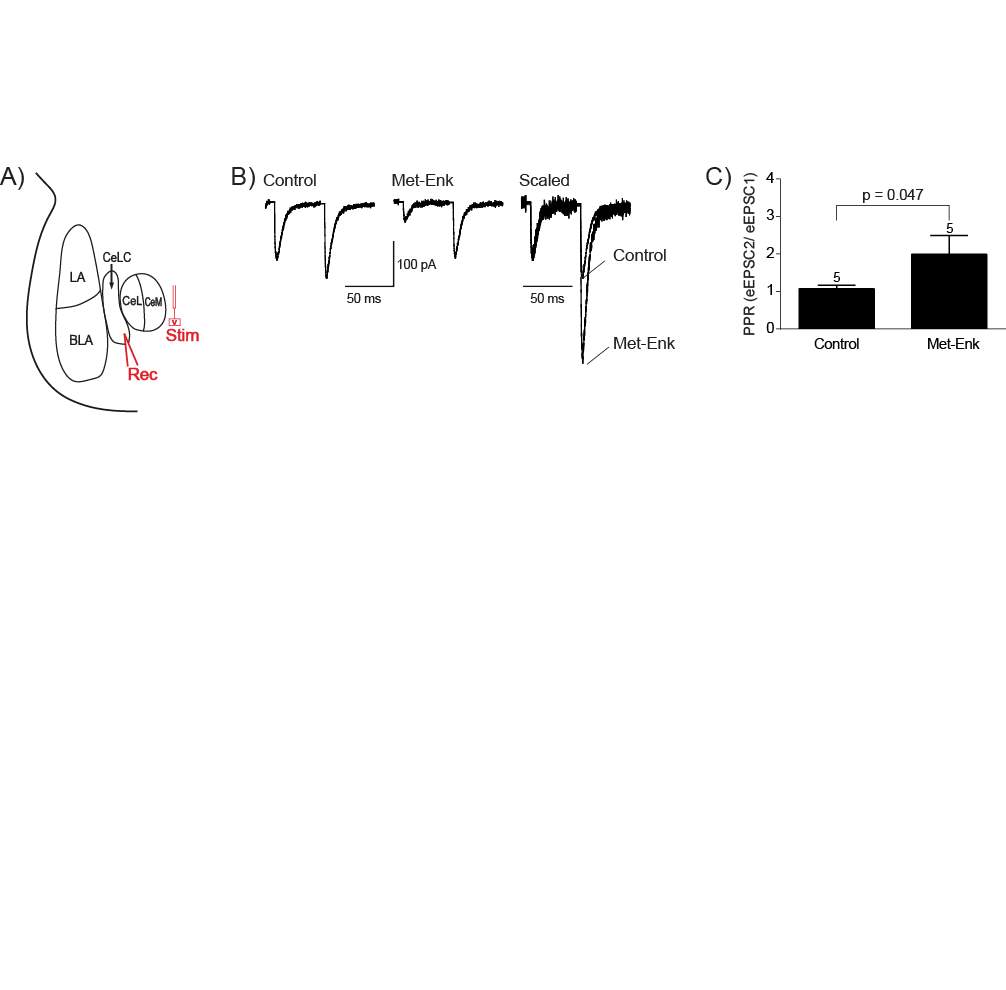
To stimulate the parabrachial synaptic inputs to the CeLC, I placed stimulating electrodes dorsomedial to the CeA (Bernard et al., 1993, Han and Neugebauer, 2004) (Figure 10A). I then made whole-cell patch clamp recordings from CeLC neurons and recorded the evoked excitatory post-synaptic current (eEPSC) at -70 mV. Idelivered two consecutive stimuli with a 50 ms interval and isolated the eEPSC by inclusion of the GABAA receptor antagonist picrotoxin (100μM) in the superfusate. The endogenous opioid Met-Enk (10µM) inhibited the first eEPSC amplitude by 56.38 ± 3.8% (n = 5). The paired pulse ratio (PPR) was calculated by dividing the second eEPSC amplitude by the amplitude of the first (eEPSC2/eEPSC1). Met-Enk significantly increased the PPR (Figure 10B & C). This suggests that Met-Enk reduces the eEPSC amplitude through acting pre-synaptically to reduce glutamate release probability.
Figure 10: Met-Enk inhibits the PB-CeLC synapse through a presynaptic mechanism.
A) Schematic diagram of stimulation and recording site. Stimulating electrodes were placed dorsomedial to the CeA to stimulate parabrachial fibers. The response of the CeLC neurons to this stimulation was recorded.
B) Example traces of eEPSCs of a CeLC neuron in control, the reduction of eEPSC amplitude in Met-Enk (10μM) and scaled to the peak amplitude of eEPSC1 of control to demonstrate the increase in PPR.
C) Bar chart of Met-Enk induced increase in PPR. Statistical significance was tested using a Paired Students T-test, one-tailed. Bar chart shows mean ± s.e.m.
4.3.2 Met-enk acts on the MOR and DORs to inhibit the PB-CeLC synapse.
Met-Enk has similar affinity for the MOR and DOR (Raynor et al., 1994). To determine whether Met-Enk inhibits glutamate release at the PB-CeLC synapse through MOR or DOR I tested whether the Met-Enk inhibition was reversed by the selective MOR antagonist CTAP (1μM) and/or the selective DOR antagonist ICI -174, 864 (ICI, 1μM). In a subpopulation of neurons both the MOR and DOR opioid receptor antagonists were required to completely reverse the Met-Enk inhibition of the PB-CeLC eEPSC amplitude (3 of 5 neurons, Figure 11A, C). CTAP completely reversed the Met-Enk inhibition in the other neurons (2 of 5 neurons, Figure 11B, C).
I also determined whether activation of the KOR regulates the PB-CeLC synapse. Superfusion of the selective KOR agonist U-69593 (U69, 300nM) did not inhibit the eEPSC amplitude or alter the PPR at this synapse (percentage inhibition of eEPSC amplitude -2.43 ± 2.8 %, control PPR: 1.60 ± 0.24, U69: 1.54 ± 0.27, p = 0.16, n = 5, paired one-tailed student’s t test) (Figure 11D).
Therefore, Met-Enk inhibits the amplitude of eEPSC at the PB-CeLC synapse through its actions at either the MOR alone or a combination of the MOR and DOR. Whereas activation of the KOR does not regulate the PB-CeLC synapse (Table 1).
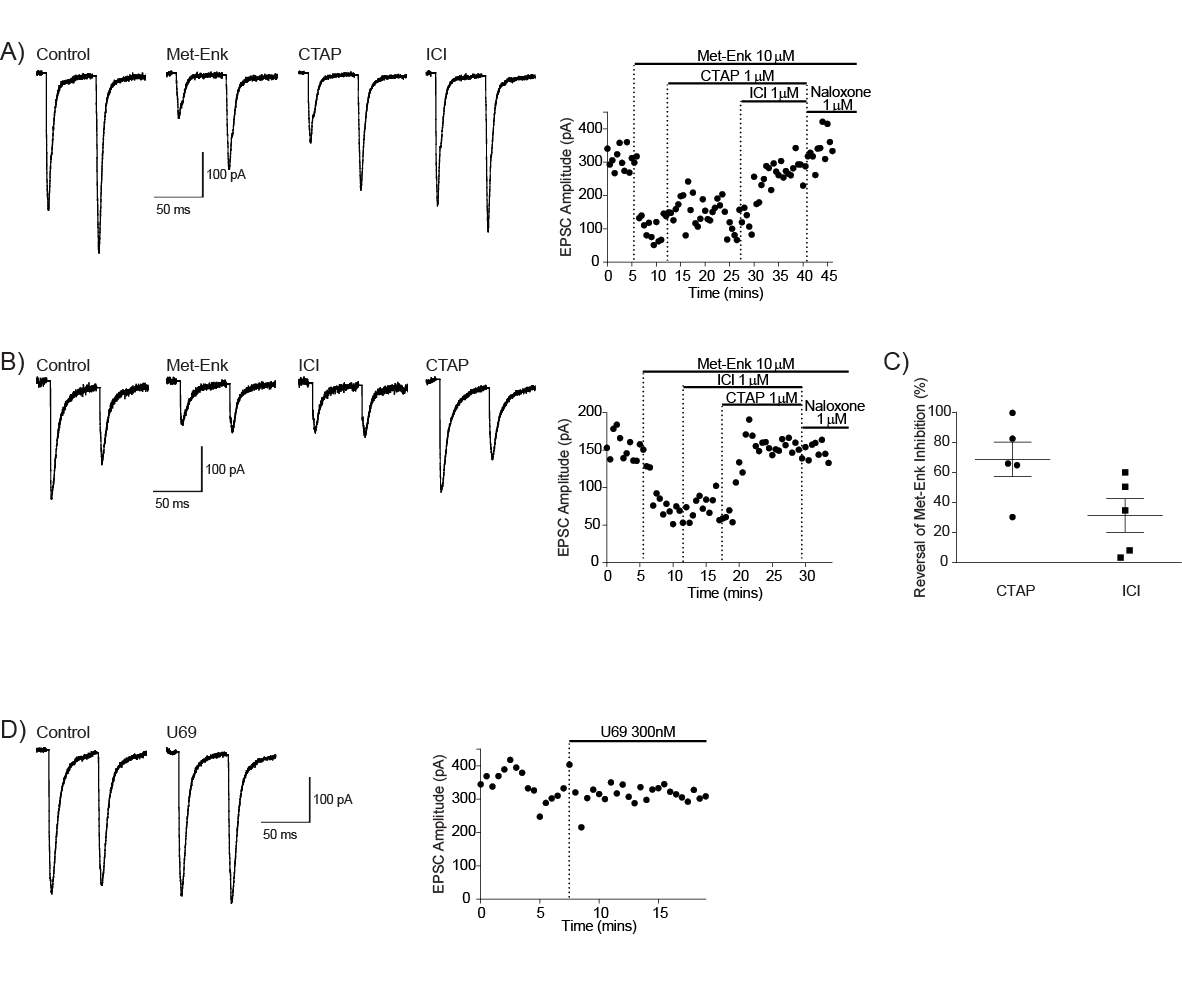
Figure 11: Met-Enk acts on MOR and DORs to inhibit the PB-CeLC synapse.
A) Example traces of eEPSC of a CeLC neuron and the time course of Met-Enk inhibition of eEPSC amplitude and reversal by selective antagonists. CTAP and ICI are both needed to reverse Met-Enk inhibition of eEPSC amplitude.
B) Example traces of eEPSC of a different CeLC neuron and the time course of Met-Enk inhibition of eEPSC amplitude and reversal by selective antagonists. CTAP completely reverses Met-Enk inhibition of eEPSC amplitude.
C) Scatter plot of the reversal of Met-Enk inhibition of eEPSC amplitude by CTAP and ICI. Each point represents an individual neuron. Graph also shows mean ± s.e.m.
D) Example traces of eEPSC of a CeLC neuron and time course in control and in U-69593 (300nM). eEPSC amplitude is not affected by U-69593.
4.3.3 Met-Enk inhibits the BLA-CeLC synapse through presynaptic MOR
To stimulate the BLA synaptic inputs to the CeLC, stimulating electrodes were placed in the BLA (Figure 12A). Application of Met-Enk (10µM) inhibited the eEPSC amplitude by 34.45 ± 4.55% (n =4). Met-Enk also significantly increased the PPR (Figure 12B & C). Thus, like the PB-CeLC synapse, Met-Enk reduces eEPSC amplitude at the BLA-CeLC synapse by reducing the glutamate release probability. Met-Enk inhibition of eEPSC amplitude at the BLA-CeLC synapse was entirely reversed by CTAP (1μM) whilst ICI was without effect (Figure 12D & E).
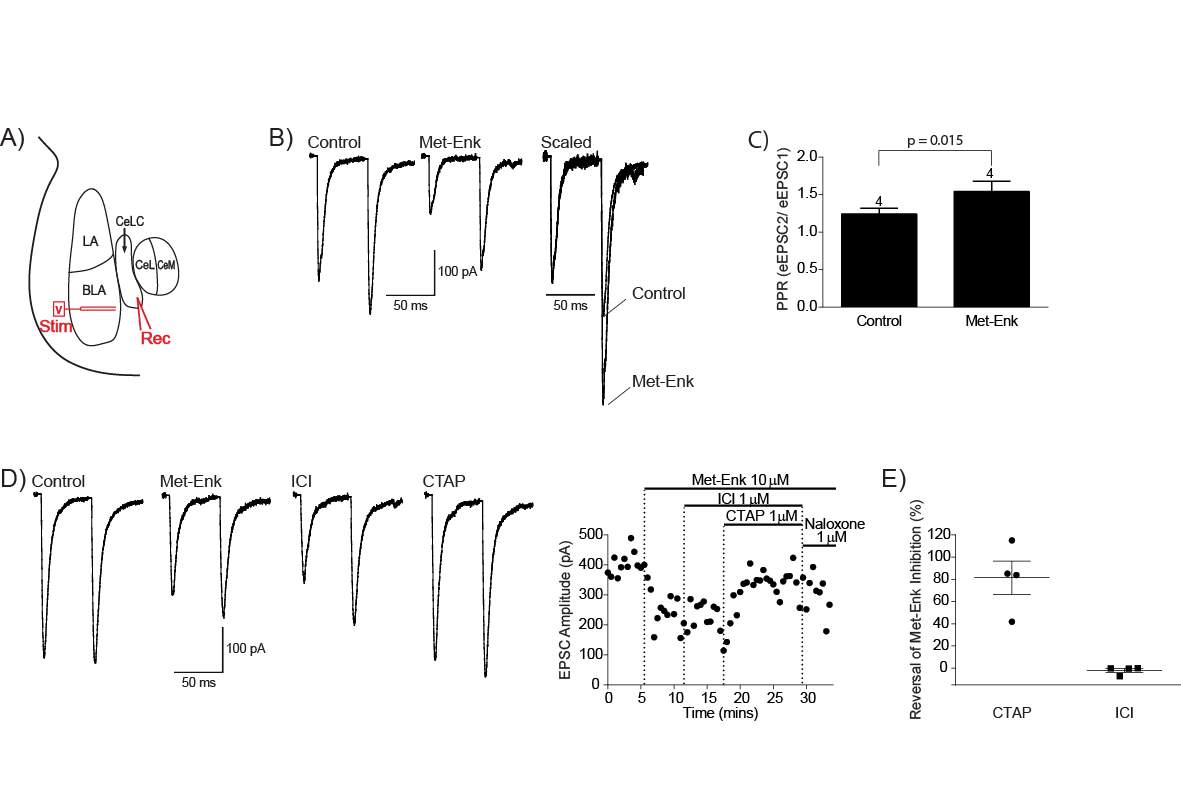
Figure 12: Met-Enk inhibits the BLA-CeLC synapse through presynaptic MOR.
A) Schematic diagram of stimulation and recording site. Stimulating electrodes were placed in the BLA to stimulate BLA fibers. The response of the CeLC neurons to this stimulation was recorded.
B) Example traces of eEPSCs of a CeLC neuron in control, the reduction of eEPSC amplitude in Met-Enk (10μM) and scaled to the peak amplitude of eEPSC1 of control to demonstrate the increase in PPR.
C) Bar chart of Met-Enk induced increase in PPR. Statistical significance tested using paired Students T-test, one-tailed. Bar chart shows mean ± s.e.m.
D) Example traces of eEPSC of a CeLC neuron and the time course of Met-Enk inhibition of eEPSC amplitude and reversal by selective antagonists. CTAP (1μM) completely reverses the Met-Enk inhibition of eEPSC amplitude.
E) Scatter plot of the reversal of Met-Enk inhibition of eEPSC amplitude by CTAP and ICI. Each point represents an individual neuron. Graph also shows mean ± s.e.m.
4.3.4 KOR modulates the BLA-CeLC synapse
The KOR agonist U69 (300nM) inhibited eEPSC amplitude by 42% in a subpopulation of neurons (2 of 5 neurons, Figure 13B). This was reversed by the KOR antagonist Nor-Binaltorphimine (Nor-BNI, 100nM, Figure 13A). U69 had no effect on eEPSC in the other neurons (3 of 5 neurons, Figure 10B).
Therefore, Met-Enk inhibits the amplitude of eEPSC at the BLA-CeLC synapse through its actions at the MOR. Distinct from the PB-CeLC synapse, activation of the KOR also inhibits the BLA-CeLC synapse in a subpopulation of neurons (Table 1).
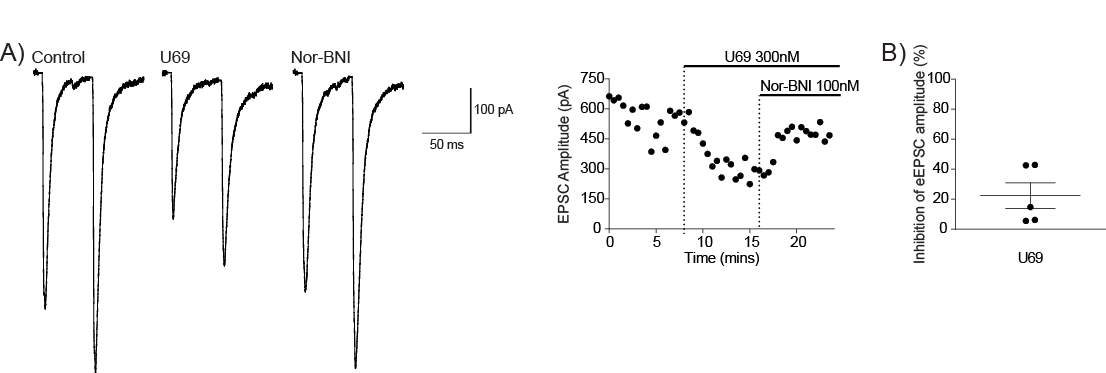
Figure 13: KOR modulates the BLA-CeLC synapse.
A) Example traces of eEPSC of a CeLC neuron and the time course of U69 inhibition of eEPSC amplitude and reversal by the selective κ antagonist Nor-BNI.
(B) Scatter plot of U69 inhibition of eEPSC amplitude. Each point represents an individual neuron. Graph also shows mean ± s.e.m.
Table 1: Opioids differentially regulate activity at the PB-CeLC and BLA-CeLC synapse.
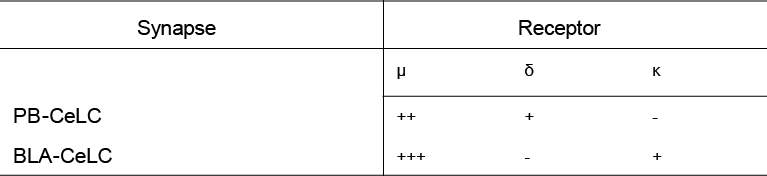
The different levels of opioid regulation at the PB-CeLC and BLA-CeLC synapse. + indicates level of receptor activity. The MOR is responsible for most opioid regulation at both synapses. There is small amount of DOR mediated regulation at the PB-CeLC synapse and a small amount of KOR mediated regulation at the BLA-CeLC synapse.
4.4 Discussion
The present study investigated opioid regulation of the PB-CeLC and BLA-CeLC synapses. I found that opioids inhibit both synapses through a presynaptic mechanism, however the receptors responsible differs between the two synapses. Opioid inhibition at the PB-CeLC synapse is through MOR and DOR activation, whereas at the BLA-CeLC synapse, opioid inhibition is through MOR and KOR activation (Table 1).
Parabrachial inputs to the CeLC are important for forming the association between a painful stimulus and the environment in which it occurs (Sato et al., 2015, Watabe et al., 2013, Han et al., 2015). This associative learning is important in a patient’s handling of pain, as they may avoid or restrict certain activities because of the painful experience (Vlaeyen, 2015). MOR produces its analgesic effects by reducing the emotional and aversive component of pain (Zhang et al., 2013, Price et al., 1985, Kupers et al., 1991, LaGraize et al., 2006). Much of the opioid inhibition at the PB-CeLC synapse was through MOR, with a subpopulation of neurons with some DOR inhibition. MOR also inhibits BLA inputs to the CeLC, which provides polymodal sensory information to the CeLC (Moga et al., 1995, Vertes and Hoover, 2008, Turner and Herkenham, 1991, McDonald et al., 1996, LeDoux et al., 1990b, Doron and Ledoux, 1999), information that is important for associative learning. Inhibition of glutamate release from PB and BLA inputs to CeLC by MOR, could block the associated learning involved in pain affect, thereby providing a mechanism of MOR inhibition of the affective component of pain. This MOR mediated inhibition of pain affect is seen in the ACC a region implicated in the generation of the emotional unpleasantness of pain (Rainville et al., 1997, Johansen et al., 2001) , where injection of morphine into the ACC, attenuates pain affect (LaGraize et al., 2006). The CeLC receives inputs directly from the ACC (McDonald et al., 1996) and indirectly via the BLA (McDonald et al., 1996). It also sends inputs to the ACC via the substantia innominata (Bourgeais et al., 2001). The CeLC’s modulation of pain affect could involve the integration of information on pain unpleasantness from the ACC, with the sensory information it receives from the PB and BLA to produce the association between the emotional unpleasantness of pain with the environment it occurs in. MOR mediated regulation of pain affect could thus involve parallel inhibition of the PB-CeLC synapse and ACC leading to reduced associative learning and generation of pain unpleasantness. However, given the reciprocal connections between the CeLC and ACC, MOR regulation may also involve feedforward inhibition of the two regions.
DOR’s role in pain analgesia is more complex than MOR. DOR is mostly found intracellularly in the cytoplasm (Arvidsson et al., 1995a, Cahill et al., 2001a, Cheng et al., 1997) and this is thought to be the reason why some areas such as the PAG, even though it has relatively high expression of the DOR protein, does not have DOR mediated activity (Connor et al., 1999, Chieng and Christie, 1994, Hack et al., 2005). DOR is thought to only have activity when certain conditions such as chronic morphine treatment (Chieng and Christie, 2009, Cahill et al., 2001b) or chronic inflammatory injury (Cahill et al., 2003) cause trafficking of the receptor to the plasma membrane (Cahill et al., 2003, Cahill et al., 2001b, Chieng and Christie, 2009). In line with this, DOR agonists have better analgesic properties in chronic pain conditions than acute pain (Cahill et al., 2003, Mika et al., 2001), presumably because they undergo trafficking in chronic pain conditions. My findings on DOR modulation of the PB-CeLC and BLA-CeLC synapse provides a different perspective on how DOR modulates synapses in normal physiology. Despite the high expression of DOR (Le Merrer et al., 2009, Erbs et al., 2015) and it’s mRNA (Mansour et al., 1995, Mansour et al., 1994, Poulin et al., 2006, Le Merrer et al., 2009) in the BLA, there was no DOR mediated regulation at the BLA-CeLC synapse. This fits with the school of thought of the DOR not having activity in normal circumstances. However, the PB-CeLC synapse does have DOR modulation. Admittedly the PB only has light labelling of DOR (Arvidsson et al., 1995a, Mansour et al., 1995, Erbs et al., 2015) and it’s mRNA (Mansour et al., 1994) and PB neurons are not inhibited by DOR activation (Christie and North, 1988). Thus, the DOR modulation seen at the PB-CeLC could be due to stimulation of non-PB fibers. Nevertheless, this result does show that it is possible to have DOR modulation in normal circumstances. This may be because DOR can be trafficked to terminals where it can modulate synapses, however in the soma, it is not readily trafficked onto the plasma membrane. This ability to traffic to terminal endings appears to be synapse dependent because of the difference between the BLA-CeLC and PB-CeLC synapse. This is also made more evident by recent evidence that the BLA-ITC synapse is modulated by DOR under control conditions (Winters et al., 2017).
The KOR has even less of a presence in the pain analgesic than DOR. KOR causes dysphoria and aversion in the CNS (Land et al., 2008, McLaughlin et al., 2003, Van’t Veer and Carlezon, 2013, Mucha and Herz, 1985, Funada et al., 1993), hence its use in pain treatment is restricted to the periphery. The findings from this study may have indicated a synapse contributing to KOR mediated dysphoria and aversion. Interestingly behavioural studies have revealed an involvement of the BLA in KOR mediated aversion. Microfusion of KOR antagonist into the BLA reduces fear potentiated startle (Knoll et al., 2011) and can also attenuate KOR agonist induced anxiety-like behavior in animals (Smith et al., 2012). It is not known exactly what synapse KOR is acting on, however given that, KOR regulates the BLA-CeLC synapse in a subpopulation of neurons, KOR induced aversion in the BLA may be due to inhibition of the BLA-CeLC synapse.
CeLC neurons receive nociceptive information from the PB (Bernard et al., 1993, Bernard et al., 1992, Bester et al., 1997) and polymodal sensory information from the BLA (Sah et al., 2003, Marek et al., 2013, Pape and Pare, 2010, Neugebauer, 2015). The two inputs provide CeLC neurons with information to form the association between a painful stimulus and the environment. Both the PB and BLA inputs are glutaminergic (Dong et al., 2010) and are inhibited by opioids. CeLC neurons also expressed opioid receptors postsynaptically and are inhibited by MOR through activation of GIRKS (Chieng et al., 2006), inhibiting their projection to areas such as the substantia dorsalis innomimata (Bourgeais et al., 2001) and in turn the relay of nociceptive information to areas that encode pain affect. Morphine, the most commonly administered opioid acts at MOR (Matthes et al., 1996, Sora et al., 1997b, Tian et al., 1997, Loh et al., 1998) and both these inputs have most of their opioid regulation through this receptor. Thus, when a person is given morphine, it could be inhibiting PB and BLA inputs to the CeLC and thus reducing the associative learning required for pain induced aversion.
Chapter 5: Optogenetic dissection of opioid regulation at the PB-CeLC synapse
5.1 Introduction
In the previous chapter, I found that opioids modulate the PB-CeLC synapse. Opioid modulation was through a presynaptic inhibition of glutamate release and acts mostly through the MOR. This finding is important as opioids particularly those that act on the MOR act by reducing the emotional unpleasantness of pain (Zhang et al., 2013, Price et al., 1985, Kupers et al., 1991, LaGraize et al., 2006). Thus, inhibition of this synapse by MOR could be the mechanism behind opioid inhibition of pain affect. I also found in the previous chapter that a small population of neurons at the PB-CeLC synapse had DOR regulation. This disagrees with previous research, which did not see DOR activity in the PB (Christie and North, 1988). The lateral PB itself has only light labelling of DOR (Arvidsson et al., 1995a, Mansour et al., 1995, Erbs et al., 2015) and mRNA (Mansour et al., 1994). Thus, I wondered whether this finding was because of stimulation of non-PB DOR positive fibers. Electrical stimulation of the PB-CeLC synapse is relatively selective and reproducible because of the visibility of the fiber tracts (Han and Neugebauer, 2004, Bernard et al., 1993), however it is still possible to recruit non-PB fibers when stimulating, especially at high voltages. A technique such as optogenetics can be used to selectively activate PB-CeLC synapse.
Optogenetics uses opsin genes such as Channelrhodopsin-2 (ChR2) to selectively activate neurons (Yizhar et al., 2011, Nagel et al., 2005, Boyden et al., 2005). Despite the advantage of using optogenetics over electrical stimulation, it does have its caveats. Introduction of ChR2 into the neurons results in changes to the normal physiology of neurons and synapses (Yizhar et al., 2011, Jackman et al., 2014, Cruikshank et al., 2010). The changes are a depression of evoked EPSCs during consecutive light stimulation in ChR2 animals (Yizhar et al., 2011, Jackman et al., 2014, Cruikshank et al., 2010). This differs from the facilitation of responses that occurs with consecutive electrical stimuli in naïve animals (Yizhar et al., 2011, Jackman et al., 2014, Cruikshank et al., 2010). The magnitude of changes is not consistent between different neurons and synapses (Yizhar et al., 2011, Jackman et al., 2014, Cruikshank et al., 2010) and also depends on the method of ChR2 delivery (Jackman et al., 2014). Light evoked EPSCs in ChR2 animals at the Purkinje cells (PC) projections onto deep cerebellar nuclei do not differ from electrically evoked EPSCs in naïve animals (Jackman et al., 2014). Whereas the facilitation of electrically evoked EPSCs at the CA3-CA1 synapse in naïve animals is reduced when light is used to evoke responses in ChR2 animals (Jackman et al., 2014).
This depression of light evoked EPSCs in ChR2 animals is possibly due to the use of AAV vectors, as light evoked responses from ChR2 animals that are expressed using a non-virus such as Cre-dependent reporters and drivers do not differ from electrically evoked responses in naïve animals (Jackman et al., 2014). Furthermore, even the electrically evoked responses in AAV delivered ChR2 animals differ from naïve animals, suggesting that it is the AAV virus that changes the physiology of the synapse (Jackman et al., 2014).
As mentioned above, optogenetics can be used to selectively activate the PB-CeLC. Thus, the first aim of this study was to use optogenetics to selectively activate the PB-CeLC synapse and investigate the opioid modulation and whether it differs from electrical stimulation. The changes produced by optogenetics vary across synapses and neurons and thus the second aim of this study was to investigate whether optogenetics alters the normal physiology at the PB-CeLC synapse.
I used viral delivery to express ChR2 in the cell bodies of the lateral PB and consequently the terminals. Light illumination over the terminals originating from the PB elicits a downstream postsynaptic response in the neurons of the CeLC, which can be recorded. I found that opioid modulation of PB-CeLC using optogenetics differed from the previous chapter. Opioid modulation was through the MOR only. There was no DOR activity when the PB-CeLC synapse was selectively activated. This supports the initial hypothesis that electrical stimulation recruited non-PB DOR positive fibers. However, I also found that optogenetics alters the PPR ratio at this synapse, therefore questioning the reliability of using optogenetics as a tool to ascertain synaptic characteristics.
5.2 Aims
- To use optogenetics to selectively activate the PB-CeLC synapse and investigate the opioid regulation.
- To characterize the effects of optogenetics application at this synapse.
5.3 Results
5.3.1 Injection spread and reliability
I injected 33 animals with AAV5-HSYN-ChR2 (H134R)-EYFP viral construct and of those, 18 had expression of ChR2/EYFP in the target region. Expression was visualised using a fluorescent microscope for acute slices and a confocal microscope for fixed tissues. Injections were targeted at the external lateral portion of the PB (PBel) as this is the region, that sends nociceptive inputs to the CeLC (Bernard et al., 1993, Bernard et al., 1992, Bester et al., 1997) (Figure 14A). Most cell body expression was in the outer PBel (Figure 14B). There was some cell body expression in the central lateral and dorsal lateral PB (Figure 14B). No cell body expression was seen in the medial PB. The coordinates used were reproducible between animals within the same weight range; only 4/33 surgeries had their injection site in the wrong region of the PB. Overall ChR2/EYFP cell body expression correlated very well with terminal expression in the CeLC. There was only one instance where there was ChR2/EYFP cell body expression in the PB, but no terminal expression in the CeLC. I found that both sides of the PBel had to have expression for there to be sufficient terminal expression in the CeLC.
Virus volumes of 300nl and 500nl produced good ChR2/EYFP cell body expression and terminal expression. The spread of virus to non-target regions was the same for both volumes; however, 500nl had slightly more ChR2/EYFP terminal expression in the CeLC, suggesting that the higher volume produced slightly more cell body expression in the PBel. Cell body expression was seen in acute slices after 3 weeks in the PB, however terminal expression in the CeLC was only seen after 6 weeks. CeLC neurons only responded to light activation if EYFP/ChR2 expression could be seen under a fluorescent microscope on acute slices.
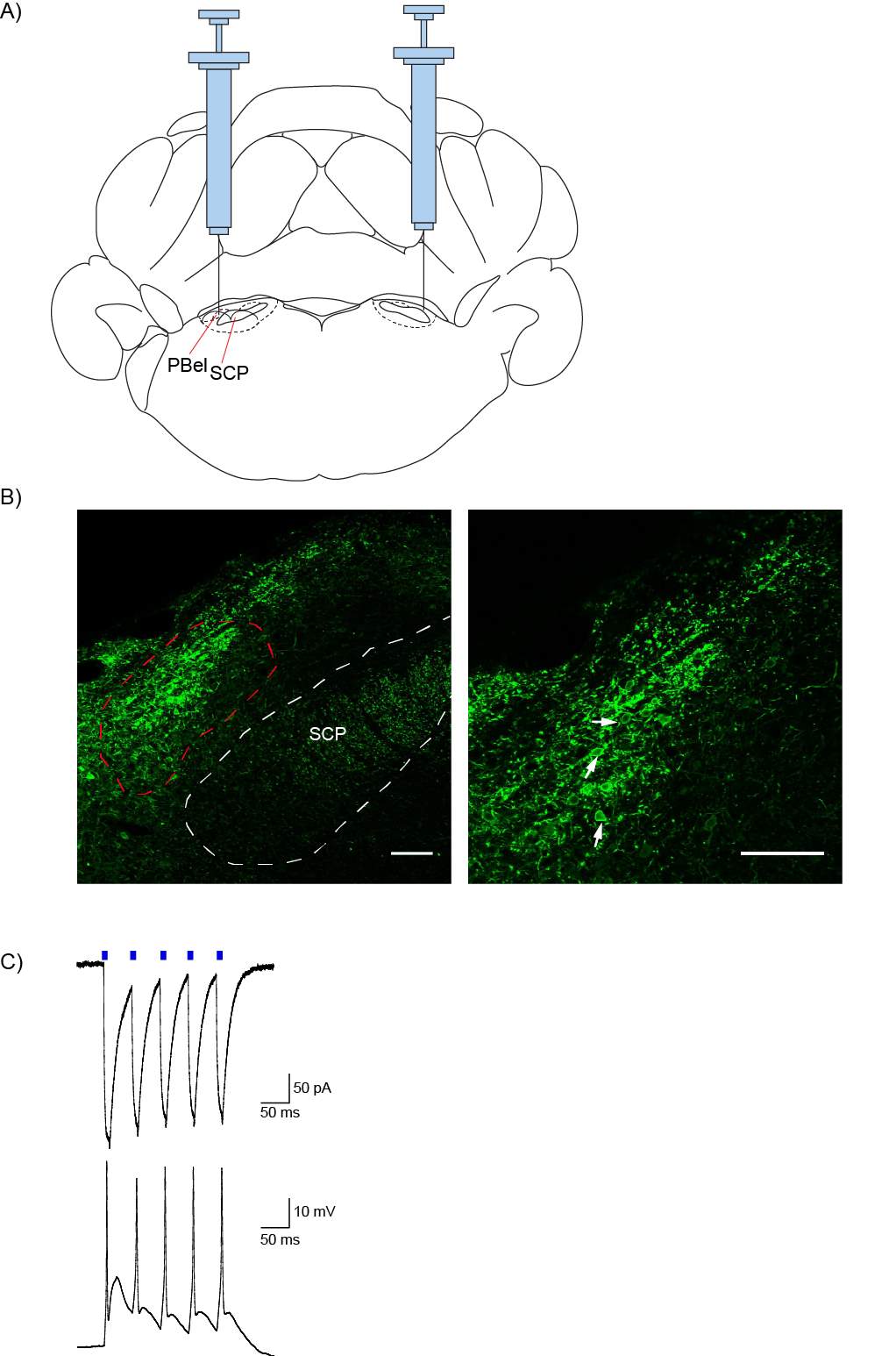
Figure 14: AAV5 injection into the PBel produces ChR2/EYFP expression in the PBel, which can be activated by blue light.
A) Coronal schematic at bregma -9.68 indicating bilateral injections of AAV5 virus to the PBel. Adapted from the brain atlas by Paxinos and Watson.
B) Low magnification (left) and higher magnification (right) confocal images of ChR2/EYFP expression in the PB. Injection was targeted at the PBel (red outline), where most ChR2/EYFP expressing cell bodies were found (white arrows). Scale bars: 100 μm. Images were taken 3 weeks after injection.
C) Light flashes (blue bars) of 10ms duration evoked inward currents (top trace, holding potential -70mV) and action potentials (bottom trace) in ChR2/EYFP expressing cells in the PBel. PBel: external lateral PB and SCP: superior cerebellar peduncle.
5.3.2 Optogenetics can be used to selectively activate the PB-CeLC synapse
ChR2/EYFP expressing neurons in the PBel responded directly to light activation. Blue light pulses of 2ms or 10ms duration produced time-locked inward currents in 8/8 PBel neurons voltage-clamped at -70 mV (current amplitude: 327.5 ± 77.6pA) (Figure 14C). Neurons with inward currents above 200pA in voltage-clamp produced action potentials when switched to current clamp (5/8) (Figure 14C). Neurons with no ChR2/EFYP expression did not respond to light activation.
Nine to twelve weeks after virus injections, there was intense expression of ChR2/EYFP terminals in the CeLC (Figure 15B). To test whether there was functional connectivity between the PBel and CeLC, CeLC neurons with soma surrounded by ChR2/EYFP boutons were recorded using whole cell patch clamp at -70mV in acute slices (Figure 15A). Picrotoxin (100μM) was perfused onto slices to block GABAA receptor mediated spontaneous activity. Light pulses of 0.1 or 0.5ms duration directly over these presynaptic boutons elicited eEPSCs in 27/34 CeLC neurons. Light evoked EPSCs were glutamatergic as they were abolished by application of the glutamate receptor antagonists NBQX (10μM) and APV (100μM) (Figure 15C & D). Evoked EPSCs had similar peak amplitudes (laser eEPSCs amplitude: 206 ± 41.11pA, n = 17 vs naïve eEPSCs amplitude: 156.9 ± 34.17pA, n = 18, p = 0.23, two-tailed Mann-Whitney test) and decay kinetics to eEPSCs stimulated by electrical stimulation in naïve animals (laser eEPSCs time constant: 4.0 ± 0.56ms, n = 7 vs naïve eEPSCs time constant: 5.3 ± 0.53, n = 11, p = 0.13, Unpaired two-tailed Students t-test). Together, these data show that optogenetics can be used to selectively activate the PB-CeLC synapse.
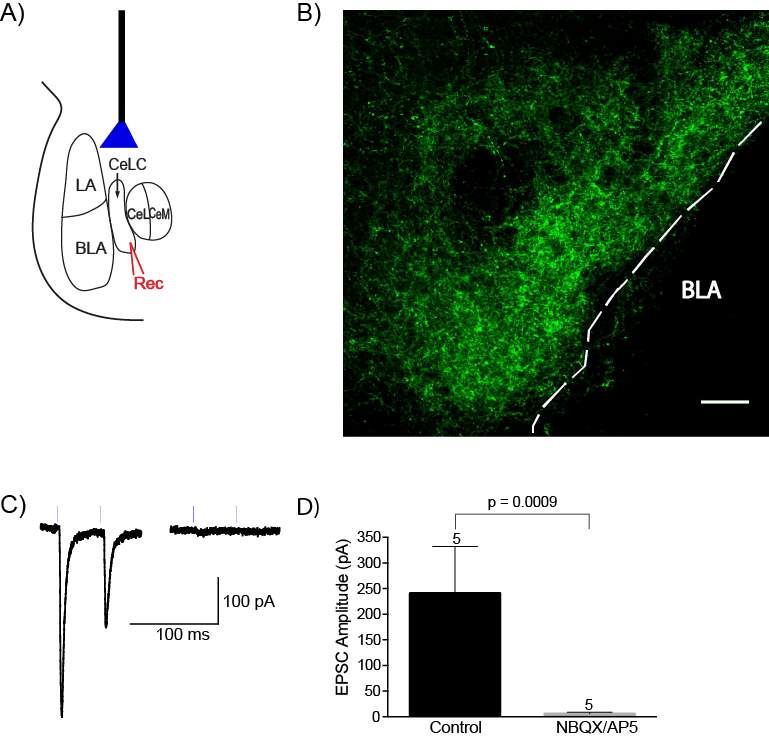
Figure 15: AAV5 injection into the PBel produces ChR2/EYFP terminal expression in the CeLC, which can be activated by light.
A) Nine to twelve weeks after viral injections, light pulses of 0.1 or 0.5ms duration was directly illuminated over ChR2/EYFP expressing presynaptic boutons in the CeLC. The response of CeLC neurons to this stimulation was recorded.
B) Confocal image of EYFP/ChR2 terminal expression in the CeLC. Scale bar: 100 µm
C) Example traces of light (blue lines) evoked EPSCs of a CeLC neuron in control (left) and after the application of NBQX (10μM) and APV (100μM).
D) Bar chart of NBQX and APV inhibition of light activated EPSCs in CeLC neurons. Statistical significance was tested using a two-tailed paired Student’s t-test. Numbers above error bars indicate number of neurons. Bar chart shows mean ± s.e.m.
5.3.3 MOR, not the DOR inhibits presynaptic glutamatergic release at the PB-CeLC synapse in ChR2 animals
In the previous chapter, opioid inhibition of the PB-CeLC synapse was through the MOR and DOR. This finding was in disagreement with a previous study, where there was no DOR activity in the lateral PB(Christie and North, 1988). To test whether this DOR inhibition was on synaptic release from non-PB fibers, I used optogenetics to selectively activate the PB-CeLC synapse. I delivered two consecutive light pulses of 0.1 or 0.5ms duration with an inter-stimulus interval of 50ms over ChR2-EYFP expressing presynaptic boutons and made whole-cell patch clamp recordings from CeLC neurons and recorded eEPSCs at -70mV. The selective MOR agonist DAMGO significantly reduced the first eEPSC amplitude (control eEPSC amplitude: 284.9 ± 140.1pA vs DAMGO eEPSC amplitude: 80.82 ± 35.3, n = 6, p = 0.031, two-tailed Wilcoxon matched-pairs signed rank test) and increased the PPR (Figure 16D &E). This was reversed by the selective MOR antagonist CTAP (Figure 16A). In contrast, the DOR agonist Deltorphin II had no significant effect on the first eEPSC amplitude (control eEPSC amplitude: 113.50 ± 46.13 vs deltorphin II eEPSC amplitude: 108.60 ± 51.82, n = 4, p = 0.62, two-tailed paired Student’s t-test) or PPR (control ppr 0.35: ± 0.072 vs deltorphin II ppr: 0.38 ± 0.088, n = 4, p = 0.32, two-tailed paired student’s t-test) (Figure 16B & C). Therefore, when ChR2 expression in PB neurons is used to selectively activate PB-CeLC synapses, opioids only inhibit glutamate release via MOR.
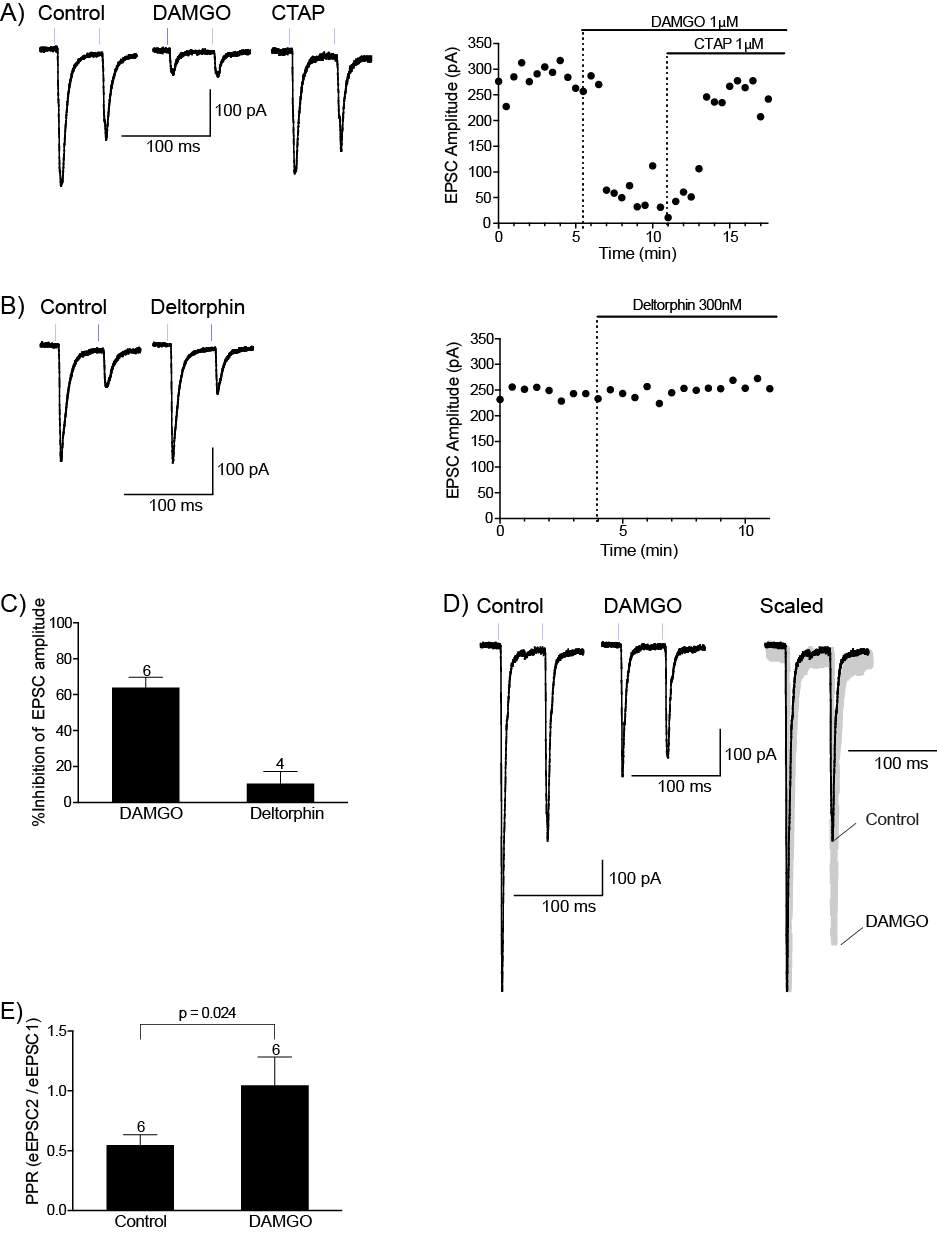
Figure 16: MOR, not the DOR inhibits presynaptic glutamate release at the PB-CeLC synapse in ChR2 animals.
A) Example traces of light activated EPSCs of a CeLC neuron and the time course of DAMGO inhibition of EPSCs amplitude and reversal by the selective antagonist CTAP.
B) Example traces of a CeLC neuron and time course in control and in Deltorphin. Light activated EPSCs are not affected by Deltorphin.
C) Bar chart of DAMGO and Deltorphin inhibition of light activated EPSC amplitude.
D) Example traces of light activated EPSCs of a CeLC neuron in control, the reduction of EPSCs amplitude in DAMGO and scaled to the peak amplitude of eEPSC1 of control to demonstrate the increase in PPR.
E) Bar chart of DAMGO induced increase in PPR. Statistical significance was tested using a two-tailed paired student’s t-test. Numbers above error bars represent number of neurons. Bar charts show mean ± s.e.m.
5.3.4 AAV5 driven expression of ChR2 alters the PPR at the PB-CeLC synapse
Light activated currents depress with consecutive stimuli compared to electrically evoked currents in naïve animals (Jackman et al., 2014, Cruikshank et al., 2010). The depression is synapse dependent, with some synapses showing more depression than others (Jackman et al., 2014). In the previous experiments, I noticed that all the light activated EPSCs show a PPR depression (Figure 15C,16A & C). To investigate further, I delivered two consecutive light or electrical stimuli 50ms apart to compare the PPR of light activated currents in ChR2 animals with electrically evoked currents in naïve animals at the PB-CeLC synapse. There is evidence that the mechanism behind the PPR depression seen in light activated currents is due to the actual AAV vector rather than the ChR2 channel (Jackman et al., 2014). To test this at the PB-CeLC synapse, I also looked at the PPR ratio of electrically evoked currents in ChR2 animals. There was a significant difference between the three groups (F(2,36) = 35.84, p < 0.0001, one-way ANOVA) (Figure 17). Light activated currents in ChR2 animals had significantly lower PRR compared to electrically evoked currents in naïve animals (Figure 17). Interestingly PPR of electrically evoked currents in ChR2 animals was also significantly lower than electrically evoked currents in naïve animals (Figure 17B), agreeing with previous study that it is AAV virus, not the opening of ChR2 channels that alters the PPR (Jackman et al., 2014). PPR of light activated currents in ChR2 animals was not significantly different from the PPR of electrically evoked currents in ChR2 animals (Figure 17B). Together, these data show that like other synapses in the brain, the PPR of PB-CeLC synapse is altered by the AAV5 driven expression of ChR2. This may be problematic in the future use of viral injections to selectively activate this synapse, as it suggests that the synapse does not function normally in the presence of AAV. This will need to be taken into account when interpreting results obtained using AAV.
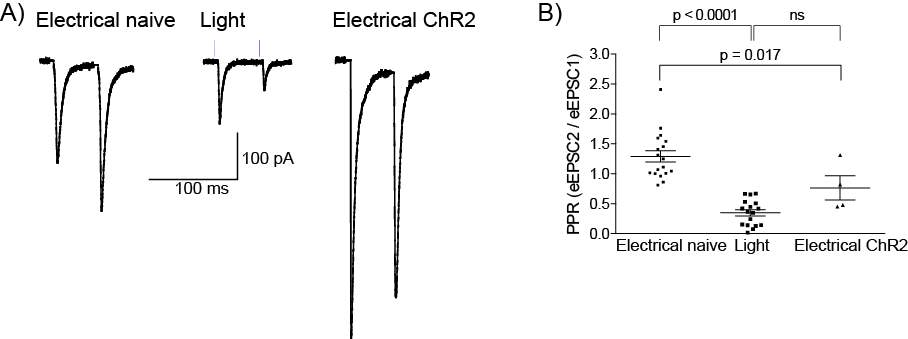
Figure 17: AAV5 driven expression of ChR2 alters the PPR ratio at the PB-CeLC synapse.
A) Example traces of electrically evoked EPSCs in naïve animals, light activated EPSCs in ChR2 and electrically evoked EPSCs in ChR2 animals.
B) Scatter plot of PPR across the three groups. Light activated currents and electrically evoked currents in ChR2 animals had significantly lower PPR compared to electrically evoked currents in naïve animals. Statistical significance was tested using a one-way ANOVA followed by Tukey’s multiple comparison tests. Dots represent individual neurons and graph also shows mean ± s.e.m.
5.4 Discussion
I used optogenetics to selectively activate the PB-CeLC synapse and investigate the opioid modulation at this synapse. I found that opioid modulation differed from the previous chapter, where electrical stimulation was used to activate this synapse. Opioid modulation was only through the MOR. I also found that PPR was lower at the PB-CeLC synapse in ChR2 animals compared to naïve animals. This may be due to the AAV vector rather than the action of ChR2.
Parabrachial fibers travel through a ventral pathway where they cross the ansa peduncularis laterally and caudally to innervate CeLC neurons (Sarhan et al., 2005). Bipolar stimulating electrodes are placed dorsomedial to the CeA to activate the fibers travelling through this pathway (Bernard et al., 1993, Han and Neugebauer, 2004, Sarhan et al., 2005). It is possible that stimulation of these inputs, also innervate non-PB inputs onto CeLC neurons, namely several mixed inputs that travel dorsal to the CeA (See Chapter 3). These mixed inputs include polymodal sensory information from the thalamus (Moga et al., 1995, Vertes and Hoover, 2008), hypothalamus (Canteras et al., 1994), entorhinal cortex (McDonald and Mascagni, 1997) and lateral occipital area (McDonald et al., 1996). They also include inputs from areas delivering affective information such as prefrontal cortex, insular cortex and anterior cingulate cortex (McDonald et al., 1996). Interestingly in chapter 3, there was a separation of results obtained when the two inputs were stimulated and compared, suggesting that placement of electrodes at the different locations activate different inputs.
Nevertheless, optogenetics offers an increased certainty on the selectivity of activation. However, the use of optogenetics come with a few caveats. First and foremost is the practicality of the technique. Stereotaxic surgeries are time-consuming and the success rate of injections can be low. In this study, only 18 out of 33 surgeries were successful. The length of time needed for ChR2 expression is also long. AAV is a single-strand DNA virus that must form its double-strand within the host cell before gene expression can take place (Aschauer et al., 2013). This is postulated to be the rate-limiting step in the expression of genes delivered via AAV (Aschauer et al., 2013, Ferrari et al., 1996), as the use of self-complementing AAVs (scAAV) in which the two halves of a single-stranded AAV genome are packaged together to form an intra-molecular double-strand, reduces the length of time needed for gene expression compared to the conventional single-strand AAVs (McCarty et al., 2001, Aschauer et al., 2013). Thus the time needed for the formation of the double-stranded DNA in conventional AAVs explains the significant length of time needed for ChR2 expression. I obtained the best ChR2 terminal expression when experiments were conducted 9 weeks post injection. This was longer than past optogenetics experiments at this synapse (Carter et al., 2013, Sato et al., 2015) however I also had a higher success rate for light evoked responses. This is presumably due to more axonal transport of the AAV/ChR2 to the axon terminals (Castle et al., 2014, Cearley and Wolfe, 2007).
Opioid modulation of PB-CeLC synapse using electrical stimulation was through MOR and DOR, whereas using light stimulation in ChR2 animals, it was only through MOR. A previous study also found no DOR mediated activity in the lateral PB (Christie and North, 1988). This previous study and the difference I saw between electrical and optogenetics, implies that electrical stimulation due to its reduced selectivity, may also stimulate non-PB inputs. This data provides stronger evidence that opioid regulation of this synapse is only through the MOR. As mentioned in the introduction, the MOR has the best antinociceptive properties of the opioid receptors and most commonly administered opioid analgesics such as morphine exert their effects through the MOR (Kieffer, 1999, Pradhan et al., 2011). Thus, as mentioned in chapter 4, MOR regulation of this synapse could be part of the mechanism of action of these opioid analgesics.
I found that there is a reduced PPR at PB-CeLC in ChR2 animals compared to naïve animals. This has also been shown at other synapses (Yizhar et al., 2011, Jackman et al., 2014, Cruikshank et al., 2010). This complicates the interpretation of the opioid result, as the mechanism behind the paired pulse depression in ChR2 animals may also affect DOR activity. There are a few possible mechanisms behind the paired pulse depression. ChR2 desensitizes and takes about 25 seconds to recover (Nagel et al., 2003, Nagel et al., 2005, Mattis et al., 2011, Lin, 2011), much longer than the 50ms inter-stimulus interval used to measure PPR. ChR2 also increases release probability by increasing activation of voltage-gated calcium channels (Zhang and Oertner, 2007). This increase in release probability would lead to paired pulse depression. Opioids inhibit neurotransmitter release in some regions by inhibiting voltage-gated calcium channels (Moises et al., 1994, Acosta and Lopez, 1999, Wu et al., 2004). Thus, if opioids inhibit neurotransmitter release through this mechanism at the PB-CeLC synapse and, there is increased activation of voltage-gated calcium channels in ChR2 animals, one would expect greater opioid activity and enhancement of DOR activity rather than a reduction. The intrinsic properties of the ChR2 channels may still be the mechanism behind the artificial paired pulse depression, however it seems unlikely that it would affect DOR activity.
Paired pulse depression may also be related to the use of AAV vectors. The depression does not occur in ChR2 experiments obtained using transgenic animals. It is also dependent on the type of AAV serotype used, as AAV5 the serotype used in this study produces this depression but AAV9 doesn’t (Jackman et al., 2014). Interestingly I showed that electrical stimulation of PB-CeLC synapse in ChR2 animals also has reduced PPR. This has been shown at other synapses (Jackman et al., 2014). This suggests that the paired pulse depression is not due the opening of ChR2 channel but rather the AAV vector itself. The mechanism behind this AAV induced depression is unknown, however AAV5 has been shown to cause reactive astrocytosis, which reduces neurotransmitter release at inhibitory synapses (Ortinski et al., 2010). While reactive astrocytosis does not affect excitatory synapses (Ortinski et al., 2010), and hence cannot explain the depression seen here and other excitatory synapses (Yizhar et al., 2011, Jackman et al., 2014, Cruikshank et al., 2010), it does show that AAV particularly AAV5 can have a detrimental effects at synapses. Whilst the mechanism behind the depression seen at this synapse is unknown, it is possible that hinders DOR activity. It would be interesting to conduct experiments in CHR2 animals and use electrical stimulation to investigate DOR activity at the PB-CeLC synapse.
The effects of optogenetics on synaptic transmission depends on viral delivery, ChR2 and on the properties of the synapse itself (Yizhar et al., 2011). The paired pulse depression seen at the PB-CeLC synapse and other synapses (Cruikshank et al., 2010, Jackman et al., 2014) was not seen at the synapse between the cerebellar purkinje cells and the deep cerebellar nuclei (PN-DCN). This study was originally set out to investigate opioid modulation at the PB-CeLC synapse using optogenetics, however it is also important, as it is the first to characterize the effects of optogenetics at this synapse. Optogenetics provide a great opportunity to elucidate the functional connectivity between synapses, however must be used with caution when attempting to characterize synapses. While transgenic animals and use of AAV9 has been shown to abolish this artificial synaptic depression at other synapses (Jackman et al., 2014), it remains to be seen whether this is the case for the PB-CeLC synapse.
Chapter 6: Calcitonin gene-related peptide as a neuromodulator of the PB-CeLC synapse
6.1 Introduction
Calcitonin gene-related peptide (CGRP) is a 37-amino acid peptide first discovered from alternative splicing of the gene for the hormone calcitonin (Russell et al., 2014, Walker et al., 2010, Doods et al., 2007). CGRP is expressed at high levels in the periphery and CNS and is a possible target in the treatment of migraines (Russell et al., 2014, Walker et al., 2010, Doods et al., 2007). CGRP is expressed at all levels of the spino-parabrachio-amygdaloid pain pathway and is used as a marker for parabrachial terminals to CeLC neurons (Carter et al., 2013, Chieng et al., 2006, Han et al., 2015, Haring et al., 1991, Kruger et al., 1988, Shimada et al., 1985). CeLC neurons also express the CGRP receptor (van Rossum et al., 1997, Oliver et al., 1998, Han et al., 2015).
CGRP terminals from the PB form asymmetric (glutamatergic) synapses with CeLC dendritic shafts and spines but also form symmetric (non-glutamatergic) synapses with CeLC soma (Dong et al., 2010, Lu et al., 2015). The presence of symmetric synaptic connection between CGRP terminals and CeLC cell bodies raises an intriguingly question of whether CGRP can directly regulate CeLC neurons. CGRP regulates synaptic transmission at the PB-CeLC synapse through a post-synaptic mechanism and increases firing of CeLC neurons in the presence of synaptic transmission (Han et al., 2010, Han et al., 2005). However, can it also directly regulate CeLC neurons in the absence of synaptic transmission? The first aim of this study was to investigate whether CGRP can modulate CeLC neurons in the absence of synaptic transmission.
The CGRP receptor has several intracellular signaling pathways (Russell et al., 2014, Walker et al., 2010). The best understood pathway is through Gαs activation of cAMP and PKA (Russell et al., 2014, Walker et al., 2010, Han et al., 2010), however there is also evidence of signaling through the Gαi/o G-protein (Disa et al., 2000, Wiley et al., 1992). The second aim of this study was to investigate whether the CGRP receptor signals through the Gαi/o G-protein at the PB-CeLC synapse by looking at whether it can regulate post-synaptic GIRK channels.
In this study, I provide preliminary data that CGRP can directly regulate CeLC neurons without synaptic transmission and that the CGRP receptor does not couple to GIRK channels.
6.2 Aims
- To determine if CGRP can directly regulate PB-CeLC synapse in the absence of fast neurotransmission.
- To investigate the signaling pathways of the CGRP receptor at the PB-CeLC synapse.
6.3 Results
6.3.1 CGRP enhances synaptic transmission at the PB-CeLC synapse through a post-synaptic mechanism
I first examined whether CGRP affected the EPSC amplitude at PB-CeLC synapse. Bipolar stimulating electrodes were placed dorsomedial to the CeA to innervate PB fibers as previously described (Figure 6Ai & 10A). I made whole-cell patch clamp recordings from CeLC neurons and recorded EPSCs at -70mV. CGRP increased EPSCs amplitude by 28.76 ± 12.9% (n = 6) (Figure 18A, B & C). I initially used the peptide CGRP receptor antagonist CGRP 8-37 to attempt to reverse the effects of CGRP, however found that it did not reverse it, in fact in one cell there was an increase in EPSC amplitude in the presence of CGRP 8-37. The CGRP 8-37 amplitude was 143 ± 19% of control (n = 3) (Figure 18A). The CGRP receptor is closely related to other peptide receptors, Adrenomedullin receptor 1 and 2 and CGRP 8-37 also has affinity for these other receptors (Doods et al., 2007, McLatchie et al., 1998, Russell et al., 2014, Walker et al., 2010). The lack of reversal by CGRP 8-37 could be due to greater affinity for Adrenomedullin receptor 1 and 2 than the CGRP receptor. The small molecule CGRP receptor antagonist BIBN4096BS (BIB) is more selective for the CGRP receptor than CGRP 8-37 (Walker et al., 2010, Salvatore et al., 2006, Hay et al., 2006, Hay et al., 2003) and thus was used in subsequent experiments. BIB reversed the effects of CGRP (reversal to 97.34 ± 12.05% of control (n = 3) (Figure 18B). These data suggest that CGRP acts on the CGRP receptor to enhance synaptic transmission at PB-CeLC synapse. A previous study found that CGRP acts through a postsynaptic mechanism to increase synaptic transmission at PB-CeLC synapse (Han et al., 2010). I next investigated whether this was the case in my hands, by using paired pulse ratio. I delivered two consecutive stimuli of equal strength with a 50ms interval to PB fibers and recorded evoked EPSCs in CeLC neurons. There was no significant difference in PPR between control and CGRP, providing no evidence for a pre-synaptic effect and therefore being consistent with CGRP acting through a post-synaptic mechanism to enhance synaptic transmission at the PB-CeLC synapse (Figure 15).
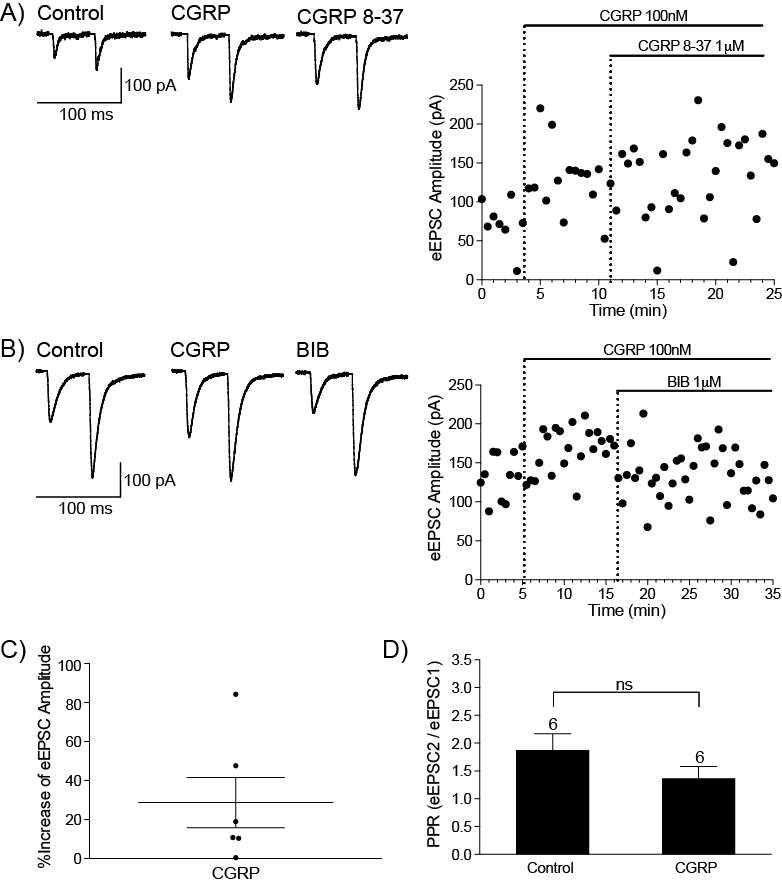
Figure 18: CGRP enhances synaptic transmission at the PB-CeLC synapse through a post-synaptic mechanism.
A) Example traces of eEPSC of a CeLC neuron and the time course of CGRP enhancement of eEPSC amplitude. The antagonist CGRP 8-37 does not reverse the effects of CGRP.
B) Example traces of eEPSC of a different CeLC neuron and the time course of CGRP enhancement of eEPSC amplitude and reversal by the antagonist BIB.
C) Scatter plot of percentage increase of eEPSC amplitude by CGRP. Each point represents an individual neuron and graph shows mean ± s.e.m.
D) Bar chart of control and CGRP PPR. Statistical significance was tested using a two-tailed Paired Student’s t-test.
6.3.2 CGRP can directly regulate CeLC neurons
The postsynaptic enhancement of synaptic transmission by CGRP could be due in part to direct cellular effects on CeLC neurons. Hence in the next set of experiments, I investigated the postsynaptic effect of CGRP on CeLC neurons. CGRP terminals form asymmetric (glutamatergic) synapses with CeLC dendritic shafts and spines, but also forms symmetric synapses (non- glutamatergic) with the soma (Dong et al., 2010, Lu et al., 2015). The synaptic contacts between CGRP terminals and CeLC cell bodies, raise the possibility that CGRP can directly regulate CeLC neurons in addition to any effects on synaptic inputs. To investigate this and the postsynaptic effect of CGRP, I tested the effect of CGRP on neuronal excitability in the presence of fast synaptic transmission and in the absence of fast synaptic transmission. Action potentials were evoked in current clamp mode by direct intracellular injections of currents of increasing magnitude (Figure 19A below). In the presence of glutamatergic and GABAergic transmission, CGRP produced an increase in action potentials in 6/6 neurons (166.59 ± 20.38% of control) (Figure 19). I then blocked glutamate receptors with the AMPA receptor inhibitor NBQX (10 µM), NMDA receptor inhibitor +/-APV (100 µM) and the GABAA receptor with Picrotoxin (100 µM) and the GABAB receptor with CGP 55845 (1 µM). When fast synaptic transmission was blocked, CGRP produced an increase in action potentials in 4/6 neurons (159.32 ± 19.49% of control) (Figure 20A & C) and a decrease in 2/6 neurons (91.67% and 71.94% of control) (Figure 20B & D). The neurons where a decrease in action potentials were observed were unlikely to be due to poor cell quality, as the series resistance and membrane potential remained stable throughout the experiment. Nor was there any noticeable differences in rostrocaudal location or intrinsic characteristics. Intriguingly the reversal of CGRP mediated effects differed between the experiments. BIB did not reverse the increase in action potentials in the presence of fast synaptic transmission (158.65 ± 18.96% of control, n = 6) suggesting that CGRP is acting on the other closely related peptide receptors. When fast synaptic transmission is blocked, BIB could reverse the increase in action potentials (116.67 ± 15.86% of control, n = 4). This indicates that CGRP can modulate CeLC neurons without fast synaptic transmission and this modulation is through the CGRP receptor.
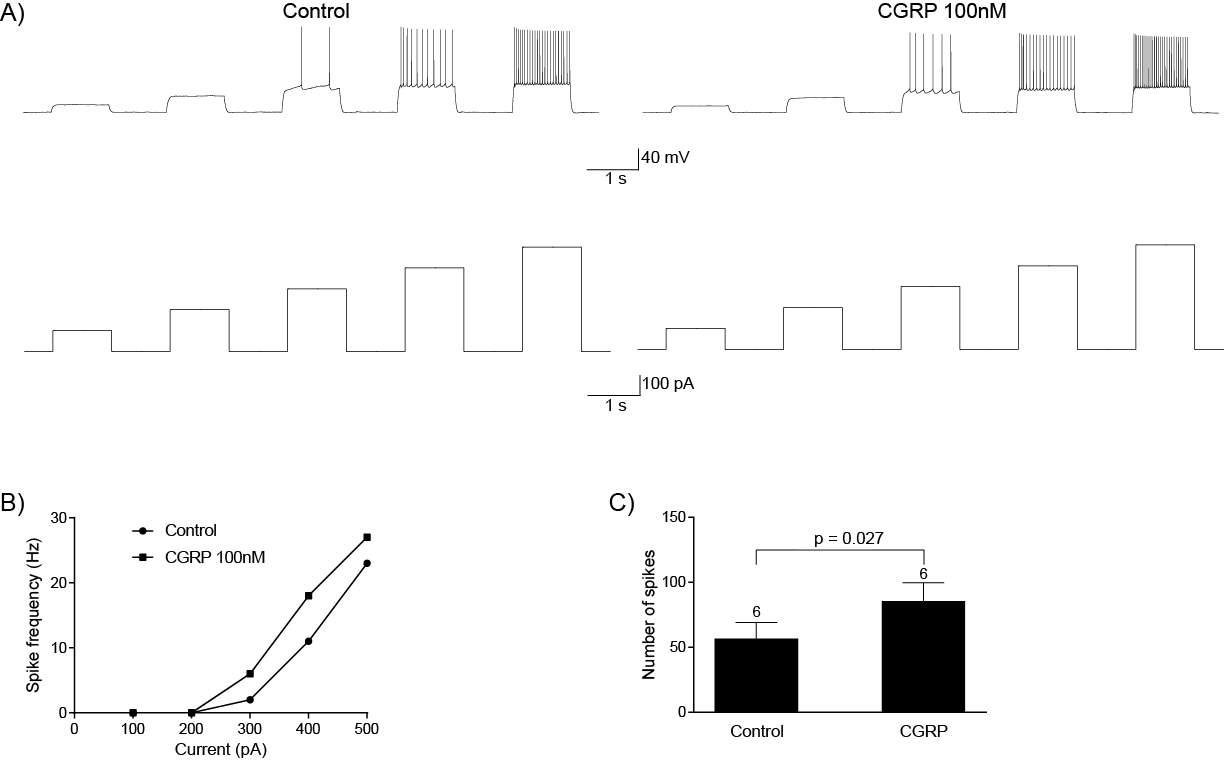
Figure 19: CGRP increases excitability of CeLC neurons in the presence of fast synaptic transmission.
A) CGRP increased the frequency of action potentials in CeLC neurons evoked by direct intracellular injections of currents of increasing magnitude in the presence of fast neurotransmission. Example traces of voltage responses (above) to increasing current injections (protocol below) under control conditions and in CGRP for an individual neuron.
B) Input/output curve of the CeLC neuron shown in A.
C) Bar chart of action potential frequency in all CeLC neurons in the presence of fast neurotransmitters. Action potential frequency is increased following application of CGRP in all neurons. Statistical significance tested using two-tailed paired Student’s t-test. Graph shows mean ± s.e.m and numbers above error bars indicate number of neurons.
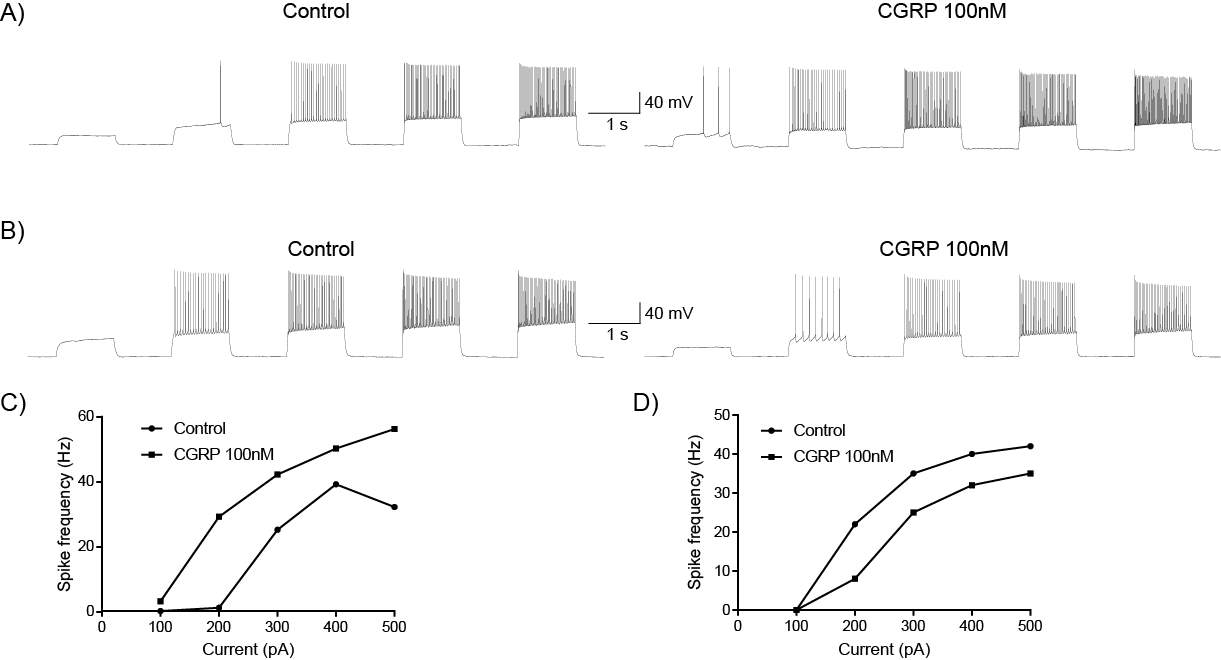
Figure 20: CGRP can modulate CeLC neuronal excitability in the absence of fast synaptic transmission.
A) CGRP increases neuronal excitability in some CeLC neurons and B) decreases neuronal excitability in another subpopulation when fast neurotransmission is blocked. The same current injection protocol was used as above.
C) Input/output curve of the CeLC neuron shown in A showing the increase in action potentials by CGRP in the absence of fast neurotransmission.
D) Input/output curve of the CeLC neuron shown in B showing the decrease in action potentials by CGRP in the absence of fast neurotransmission.
6.3.3 CGRP does not activate hyperpolarisation-activated inward currents (Ih)
Hyperpolarisation-activated inward current (Ih) is a non-selective cation channel that is activated by hyperpolarizing voltages (Banks et al., 1993, Biel et al., 2009, DiFrancesco and Tortora, 1991, Ingram and Williams, 1994, McCormick and Pape, 1990). Ih produces an inward current when cells are hyperpolarised, depolarising the cell and returning the membrane potential back to the resting membrane potential (Biel et al., 2009, Chu and Zhen, 2010, Kwak, 2012). Ih is modulated by cyclic nucleotides (Banks et al., 1993, Biel et al., 2009, DiFrancesco and Tortora, 1991, Ingram and Williams, 1994, McCormick and Pape, 1990). Agents that increase cAMP production increase Ih activation by shifting its activation to more positive voltages (Ingram and Williams, 1994, Biel et al., 2009, Banks et al., 1993, DiFrancesco and Tortora, 1991, Good et al., 2013, McCormick and Pape, 1990). Conversely agents that inhibit cAMP production, inhibit Ih by shifting activation to more hyperpolarised voltages (Biel et al., 2009, Wang et al., 2007). CeLC neurons have Ih channels (Chieng et al., 2006) thus, I hypothesized that because CGRP receptor activation increases adenylyl cyclase activity and therefore increases cAMP production through Gαs signaling (Russell et al., 2014, Walker et al., 2010), CGRP can increase Ih activation and may form the basis for the CGRP induced change in cell firing. Forskolin, a potent activator of cAMP increases amplitude of Ih currents in other brain regions (Good et al., 2013, McCormick and Pape, 1990). It was tested as a positive control to test whether cAMP activation can increase Ih currents in CeLC neurons. As mentioned above, agents that inhibit cAMP production can inhibit Ih (Biel et al., 2009, Wang et al., 2007). Opioids inhibit cAMP production through inhibition of adenylyl cyclase and can reduce Ih under control conditions (Svoboda and Lupica, 1998) or after activation of adenylyl cyclase by forskolin (Ingram and Williams, 1994). Thus, in these experiments, I also tested the effects of the MOR/DOR opioid agonist Met-Enk on Ih in CeLC neurons. To determine the effects of CGRP, forskolin and Met-Enk on Ih current, CeLC neurons were voltage-clamped at -60mV and stepped in -10mV increments to -130mV (Figure 21A). A slowly activating, voltage dependent inward current was evoked in CeLC neuorns during the hyperpolarising steps. Ih currents activated between -80mV and -100mV in CeLC neurons (n = 12). Application of CGRP did not change the amplitude of Ih currents in 3/3 cells (measured as the steady-state current minus instantaneous current) (Figure 21C). Forskolin produced an inward current of 24.58 ± 13.29 pA (n = 4) but did not affect the amplitude of Ih currents (Figure 21D). Met-Enk also did not affect the amplitude of Ih (n = 5) (Figure 21E). Ih measurements are highly sensitive to experimental conditions and cell quality (Biel et al., 2009), However, in these preliminary experiments I obtained high quality Ih recordings in 12 neurons and therefore recording quality is unlikely to be the reason for the lack of effect of CGRP, Forskolin and Met-Enk. These preliminary data suggest that Ih channels in CeLC neurons are less sensitive to cAMP activation compared to other brain regions.
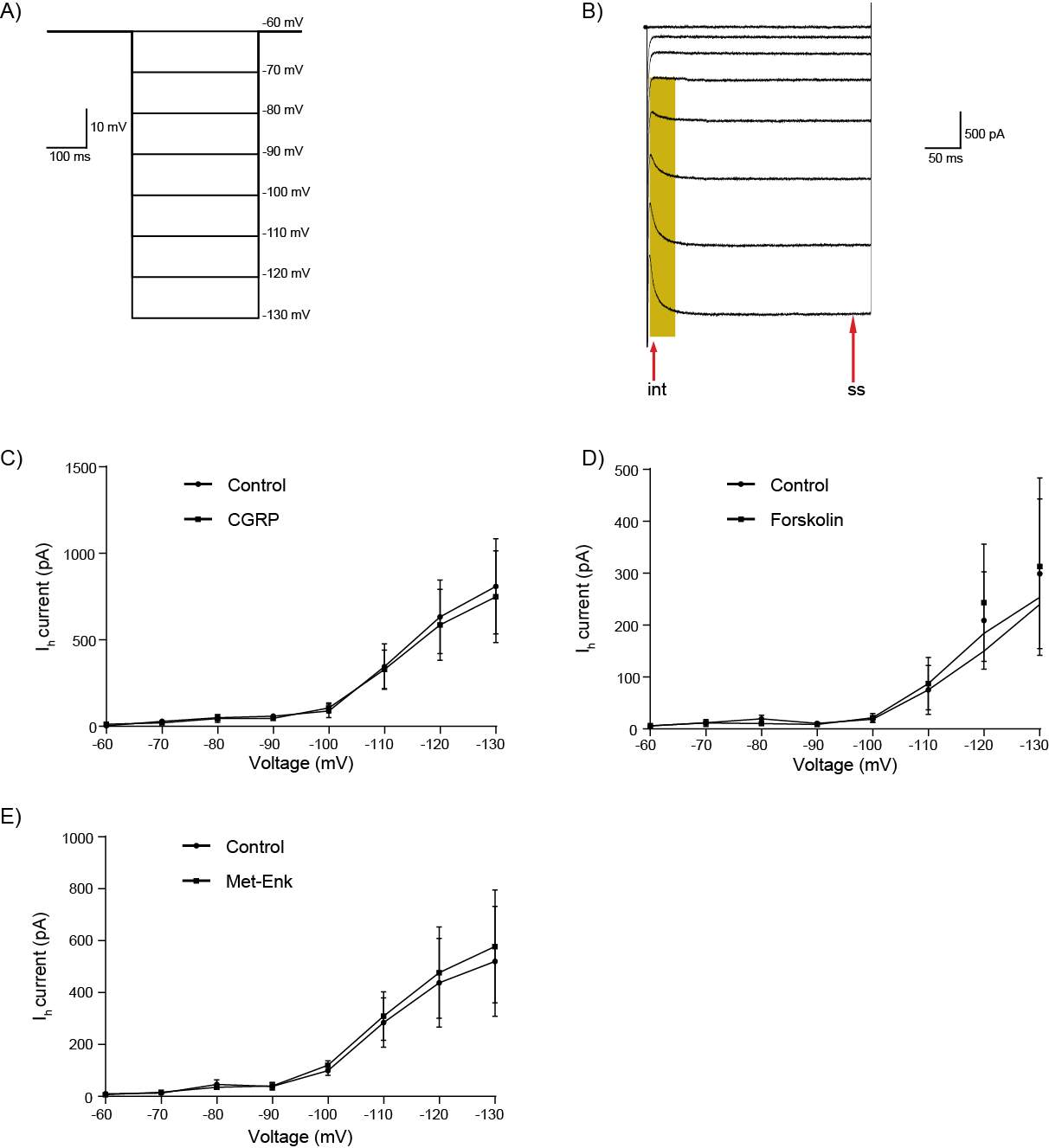
Figure 21: CGRP, Forskolin and Met-Enk do not modulate Ih channels in CeLC neurons.
A) Ih was elicited in CeLC neurons by voltage clamping at -60 mV and stepping membrane potential in -10 mV increments to -130 mV.
B) Example trace of a CeLC neuron voltage-clamped at -60 mV and stepped in -10 mV increments to -130 mV. Highlighted area indicates the slowly activating inward current characteristic of Ih current. Arrows indicate where instantaneous (int) and steady-state (ss) current were measured. Ih current amplitude was measured as the steady-state current minus the instantaneous current.
C) Grouped data of Ih currents at different voltage potentials in control and following application of CGRP (D) Forskolin and (E) Met-Enk. Graphs shows mean ± s.e.m.
6.3.4 CGRP does not activate G protein-gated inwardly rectifying potassium (GIRK) channels
The best understood signaling pathway for the CGRP receptor is through Gαs G-protein (Russell et al., 2014, Walker et al., 2010, Han et al., 2010), however there is also evidence of signaling through the Gαi/o G-protein (Disa et al., 2000, Wiley et al., 1992).G protein-gated inwardly rectifying potassium (GIRK) channels are activated by the binding of the βγ subunit of Gαi/o G-proteins, resulting in voltage-dependent outflow of K+ and hyperpolarisation of neurons (Wickman et al., 1994, Reuveny et al., 1994, Luscher and Slesinger, 2010). CGRP reduced neuronal excitability in 2 out of 6 CeLC neurons in the absence of fast neurotransmission. The reduction in neuronal excitability in these two neurons could be due to hyperpolarisation induced by GIRK channel activation. In the next set of experiments, I investigated whether the CGRP receptor in CeLC neurons can couple to Gαi/o G-proteins by examining whether CGRP can produce GIRK channel activation. I used Met-Enk which activates GIRK channels in some CeLC neurons (Chieng et al., 2006) as a positive control. Neurons were voltage-clamped at -60 mV. CGRP (100 nM) did not induce an outward current in 4/4 cells (mean current amplitude: -4.5 ± 1.7 pA) (Figure 22A). In contrast, Met-Enk (10 μM) produced an outward current in 3/5 CeLC neurons (mean current amplitude: 10.4 ± 1.5 pA) (Figure 22B), the same proportion as the previous study (Chieng et al., 2006). These preliminary data suggest that the CGRP receptor does not couple to GIRK channels in CeLC neurons and hence may not signal via Gαi/o G-protein.
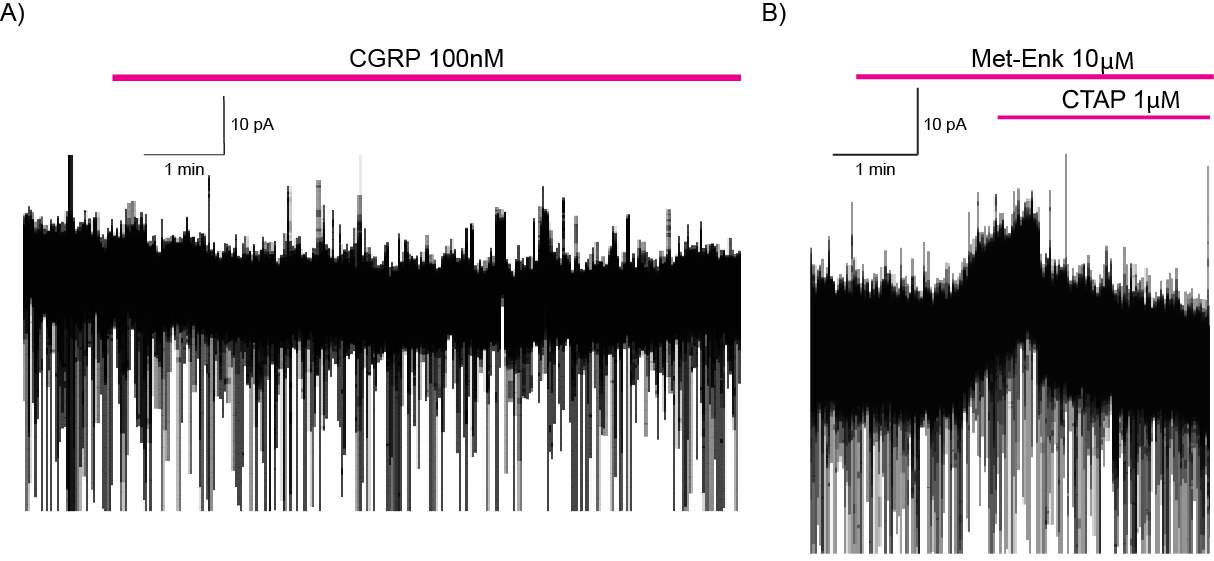
Figure 22: CGRP does not activate G protein-gated inwardly rectifying potassium (GIRK) channels.
Example traces of two different CeLC neurons voltage-clamped at -60 mV. A) Application of CGRP does not produce an outward current whereas B) application of Met-Enk results in outward current that is reversed by the MOR antagonist CTAP.
6.4 Discussion
This study determined the effects of CGRP on the PB-CeLC synapse. I found that CGRP enhances synaptic transmission through a post-synaptic mechanism. CGRP regulates the excitability of CeLC neurons in the absence of fast synaptic transmission but has no effect on post-synaptic GIRK and Ih channels.
I found that CGRP enhances synaptic transmission at the PB-CeLC synapse through a post-synaptic mechanism. This is likely through an increase in post-synaptic receptors, change in AMPA receptor subunit or change in AMPA receptor conductance. We know from my results that the PB-CeLC synapse undergoes synaptic plasticity in the absence of peripheral injury characterized by an increase in post-synaptic AMPARs. Therefore, it is possible that increased CGRP signaling after the nociceptive stimulus could boost the AMPA receptor response and therefore be responsible for the synaptic plasticity at the PB-CeLC synapse.
CGRP terminals form symmetric synapses with CeLC soma (Dong et al., 2010, Lu et al., 2015). This synaptic contact poses an interesting question on whether CGRP can regulate CeLC neurons without fast synaptic transmission. CGRP increases neuronal excitability in the presence of synaptic transmission (Han et al., 2010) however, I found that CGRP also increases CeLC neuronal excitability in the absence of glutamatergic and GABAergic transmission. One way CGRP could increase neuronal excitability without fast synaptic transmission is through Ih channels. The CGRP receptor couples to Gαs and increases cAMP production (Russell et al., 2014, Walker et al., 2010, Doods et al., 2007) which can, in turn increase Ih activation (Ingram and Williams, 1994, Biel et al., 2009, Banks et al., 1993, DiFrancesco and Tortora, 1991, Good et al., 2013, McCormick and Pape, 1990). However, both CGRP and forskolin did not affect Ih currents. This is likely because of the type of Ih channels present in the CeLC. Ih channels come from a family of channels termed hyperpolarisation activated cyclic nucleotide-gated (HCN) channels. The HCN family is comprised of four distinct members with different channel kinetics and sensitivity to cAMP (Chu and Zhen, 2010, Kwak, 2012). HCN1, the main type expressed in the CeA (Notomi and Shigemoto, 2004, Santoro et al., 2000) activates the fastest on hyperpolarisation, activating within tens of milliseconds (Santoro et al., 2000, Notomi and Shigemoto, 2004), this fits with the kinetics seen in this study. HCN1 is also the least sensitive to cAMP activation (Chu and Zhen, 2010, Kwak, 2012). If CGRP regulation of Ih is not behind the direct regulation of CeLC neuronal excitability, then a possible explanation is through activation of voltage-gated sodium ion channels. CGRP enhances tetrodotoxin (TTX) resistant sodium current in the dorsal root ganglion (Natura et al., 2005). TTX resistant sodium channels are mainly found in primary sensory neurons (Natura et al., 2005) however, a similar mechanism could be behind the increased neuronal excitability in the CeLC.
CGRP physiology is complex. A functional CGRP receptor consists of 3 components; calcitonin-receptor-like receptor (CRLR), receptor activity modifying protein-1 (RAMP1), which confers ligand specificity onto the receptor and a third component, receptor component protein (RCP) which is responsible for signal transduction (McLatchie et al., 1998, Nikitenko et al., 2006, Evans et al., 2000, Ueda et al., 2001). The CGRP receptor is closely related to other peptides in the calcitonin family, such as adrenomedullin receptor 1 and 2, with the difference been the type of RAMP complexed to the receptor (McLatchie et al., 1998, Nikitenko et al., 2006, Evans et al., 2000, Ueda et al., 2001). CGRP itself has affinity for these closely related receptors (Walker et al., 2010, Doods et al., 2007, Russell et al., 2014), making it difficult to ascertain its mechanism of action. BIB, the small molecule selective antagonist of the CGRP receptor did not reverse the CGRP increase of neuronal excitability in the presence of fast synaptic transmission, however when fast neurotransmission was blocked, BIB reversed the increase. This implies that in the presence of fast neurotransmission, CGRP acts on the other closely related receptors to increase neuronal excitability. However, when fast neurotransmission is blocked, CGRP directly increases neuronal excitability through the CGRP receptor. What this means for normal physiology is unclear, but CGRP clearly regulates CeLC neurons through more than just the CGRP receptor. These results do indicate that CGRP is capable of directly regulating CeLC neurons without fast neurotransmission, and endogenous release of CGRP could directly regulate CeLC neurons, similar to what has recently been shown with endogenous opioids (Winters et al., 2017).
This study has provided preliminary findings showing that CGRP can directly modulate CeLC neurons. This direct modulation is not through Ih or GIRK channels. CGRP enhancement of synaptic transmission at the PB-CeLC synapse will increase activity of this synapse, possibly enhancing PB-CeLC associative learning in pain. The direct modulation of CeLC neurons will increase activity of CeLC neurons, strengthening their projections and connections to areas of pain affect.
General discussion and conclusions
In the first study of this thesis, I showed that a brief nociceptive stimulus that activates the spino-parabrachio-amygdaloid pathway without damage can produce long-lasting synaptic plasticity at the PB-CeLC synapse. This brief nociceptive stimulus increased postsynaptic AMPARs at the PB-CeLC synapse for at least 3 days after the stimulus and showed similar mechanisms to long-term potentiation (LTP). This result is significant, as although the nociceptive stimulus is not comparable to a chronic pain state, it shows that plasticity can outlast a stimulus. A possible consequence of this plasticity is that the emotional response to subsequent nociceptive stimulus or injury could be enhanced, which could facilitate the formation of associative learning between pain and pain inducing activities such as sports or work. The next two studies investigated how opioids and CGRP modulate the PB-CeLC synapse. We now know that opioids inhibit glutamate release at the PB-CeLC synapse and also inhibits a subpopulation of CeLC neurons by activation of GIRK channels (Chieng et al., 2006). Opioids will inhibit nociceptive information from the PB and inhibit the CeLC output to regions of pain affect such as the ACC (Sah et al., 2003, Marek et al., 2013, Pape and Pare, 2010, Neugebauer, 2015, Rainville et al., 1997). We also know from my data, that opioids inhibit the polymodal sensory information coming from the BLA to the CeLC. Together the net effect of opioid activity may be reduced associative learning through inhibition of the PB-CeLC and BLA-CeLC synapse and reduced generation of pain unpleasantness through inhibition CeLC output to structures involved in pain affect. CGRP on the other hand, increases synaptic transmission at the PB-CeLC synapse and increases neuronal activity of CeLC neurons in the presence and absence of fast neurotransmission. The preliminary finding that CGRP can directly modulate CeLC neurons in the absence of fast neurotransmission is particularly intriguing because it alludes to an ability of CGRP to act as a neurotransmitter and facilitate an increase in CeLC activity.
7.1 Opioids and CGRP in the amygdala
It is apparent from my data that opioids and CGRP have opposing effects at the PB-CeLC synapse and in CeLC neurons. How these opposing actions affect the overall activity of the PB-CeLC synapse and CeLC neurons will presumably be determined by the peptide with the biggest influence. From my data, opioids inhibit 50-60% of the glutamate released from PB terminals and from mine and previous work, they inhibit approximately 60% of CeLC neurons through activation of GIRK channels. CGRP enhances synaptic transmission at PB-CeLC by about 30% but increases CeLC neuronal excitability in 100% of neurons tested in the presence of fast synaptic transmission and 67% of neurons tested in the absence of fast synaptic transmission. Based on this data, opioids will have a bigger influence on the PB-CeLC synapse however CGRP may have a bigger impact on CeLC output neurons. This could mean opioids will inhibit the input from the PB, reducing associative learning, however CGRP can still increase CeLC neuronal activity, thus enhancing its output to regions such as the Sld, which in turn projects to areas involved in pain affect (Bourgeais et al., 2001). The results obtained from both studies involved exogenous application of opioids and CGRP. The overall effect of these peptides will also depend on how much of these peptides are endogenously released and whether there is an interaction between the two peptides. In the CeL, CGRP terminals from the PB form axo-somatic synaptic contacts with enkephalin positive neurons (Shimada et al., 1992). This synaptic contact between CGRP terminals and CeL enkephalinergic cell bodies may facilitate the antinociceptive effect of CGRP in the CeL and CeM, where injection of CGRP increases mechanical and thermal thresholds (Xu et al., 2003). The antinociceptive effect of CGRP in the CeL and CeM is through a CGRP mediated activation of enkephalinergic CeL and CeM neurons that project to the PAG and activate the descending analgesic circuit (Xu et al., 2003). There are no synaptic contacts between CGRP terminals and enkephalin positive neurons in the CeLC (Shimada et al., 1992). This doesn’t necessarily eliminate the possibility of an interaction between opioids and CGRP in the CeLC, however unlike the rest of the CeA, intra-CeLC injections of CGRP is pro-nociceptive (Han et al., 2010), suggesting that CGRP does not influence opioid actions in the same way. What this means for CGRP as an analgesic target is unclear. Within the amygdala CGRP, depending on the region activated, can be pronociceptive and antinociceptive. Interestingly this aligns with the role of the amygdala in pain. The amygdala can also be pro-nociceptive through the PB-CeLC synapse and anti-nociceptive through activation of the descending analgesic circuit (Manning and Meyer, 1995, Borszcz, 1999, Calvino et al., 1982), raising the possibility that CGRP is involved in both these roles.
7.2 Mechanisms of pain induced plasticity
The pro-nociceptive effect of the PB-CeLC synapse in chronic pain conditions with ongoing peripheral injury also involves an increase in postsynaptic AMPARs (Ikeda et al., 2007, Cheng et al., 2011). The increase in AMPARs at the PB-CeLC synapse will increase the post-synaptic depolarization in response to release of a quanta of glutamate, ultimately leading to greater pain affect and facilitation of pain induced associative learning. In conditions with ongoing peripheral injury, continuous activation of peripheral nociceptors results in a continuous heighted activity of this synapse. However, in conditions, where there is no ongoing peripheral injury, one would expect that the increased sensitivity of the synapse would eventually return to baseline levels. However this does not appear to be the case as conditions such as back pain worsen over time even though there is no sign of a peripheral injury (Atkinson, 2004). This may be because the initial plasticity that occurs after an injury may facilitate further plasticity. Thus, chronic pain conditions with ongoing peripheral injury and conditions without ongoing peripheral injury, may share similar mechanisms. However, in the case of conditions with no ongoing peripheral drive such as chronic back pain, it is the initial plasticity that drives further changes, not ongoing activation by the peripheral injury. While the brief nociceptive stimulus I used is not representative of chronic back pain, the data suggests that the plasticity seen may be representative of the initial changes that could occur in conditions such as back pain. The plasticity produced by the brief nociceptive stimulus initially causes an increase in GluA2-lacking AMPARs, which because of their calcium permeability can act as an additional source of activity dependent calcium entry and thus facilitate subsequent synaptic plasticity (Chater and Goda, 2014, Guire et al., 2008, Plant et al., 2006, Morita et al., 2014). This data is consistent with the theory of an initial plasticity driving subsequent plasticity to cause persistent pain in conditions with no ongoing peripheral injury.
7.3 CeLC as the integrative center of the amygdala
The CeLC subdivision of the CeA is often combined with the CeL and the two are treated as a homogenous group of cells (Ciocchi et al., 2010, Haubensak et al., 2010, Delaney et al., 2007). This is because the cells in the two divisions closely resemble each other in morphology, with the main difference that the CeLC has a low cell density (McDonald, 1982). The two divisions also receive similar inputs (Moga et al., 1995, Vertes and Hoover, 2008, Canteras et al., 1994, McDonald and Mascagni, 1997, McDonald et al., 1996). Both regions receive inputs from the prefrontal cortex, insular cortex, lateral occipital area (McDonald et al., 1996), entorhinal cortex (McDonald and Mascagni, 1997), thalamus (Moga et al., 1995, Vertes and Hoover, 2008) and hypothalamus (Canteras et al., 1994). However, the inputs from the infralimbic cortex, prelimbic cortex, insular cortex (McDonald et al., 1996) and hypothalamus (Canteras et al., 1994), to the CeLC are much stronger than anywhere else in the CeA. In fact, from observation of tracing studies, within the CeA, the CeLC receives the densest inputs of all the subdivisions. The PB sends inputs to the CeL and CeLC, however it is the inputs to the CeLC that are nociceptive (Bernard et al., 1993, Bernard et al., 1992, Bester et al., 1997). As mentioned, this synapse is potentiated in various pain conditions (Fu and Neugebauer, 2008, Fu et al., 2008, Han et al., 2005, Ikeda et al., 2007, Adedoyin et al., 2010, Han and Neugebauer, 2004) and is important in associative learning and aversion in pain (Han et al., 2015; Sato et al., 2015). The CeLC is also involved in the acquisition of fear learning (Ciocchi et al., 2010) and the PB-CeLC synapse is important for important for appetite suppression in situations where it is undesirable to eat (Carter et al., 2013). Based on the many inputs of the CeLC and its various roles, I propose that the CeLC is the integrative center of the CeA and possibly the amygdala. The common theme of the various roles of the CeLC is associative learning. In pain, fear and appetite control, the CeLC due to its many inputs, is in a position to integrate incoming sensory, cognitive and affective information and provide an output to the CeM (Bourgeais et al., 2001, Jolkkonen and Pitkanen, 1998) or to Sld (Bourgeais et al., 2001). We know from my data that the CeLC is regulated by opioids and CGRP, however it is also regulated by other peptides such as neuropeptide S (Ren et al., 2013) and corticotropin-releasing factor (Fu and Neugebauer, 2008). The CeLC is important for amygdala function and further investigations should be undertaken to understand whether it differs from the CeL and also understand how the presence of various neuropeptides work together to produce its various effects in conditions such as pain, fear and appetite.
7.4 Future directions and conclusion
Chapter 4,5 and 6 established the role of opioids and CGRP in the normal physiology of the PB-CeLC synapse. It would be interesting to investigate the effects of the modulators at this synapse in animals that had been treated with the nociceptive stimulus in future studies. Opioids inhibit glutamate release at the PB-CeLC synapse however there was no evidence that the nociceptive stimulus changes neurotransmitter release at this synapse. Thus in an experiment, where opioid inhibition of AMPAR mediated response is used as the output, the effect may be a reduction in the opioid inhibition as there are more AMPARs. However, there may also be no change, since the two mechanisms may cancel each other out. Because the nociceptive stimulus does not change neurotransmitter release, it likely does not affect opioid inhibition of neurotransmitter release. Reduced release of neurotransmitter will reduce the impact of the increase in postsynaptic receptors. In a clinical sense, this is important as it means even though this synapse has undergone plasticity, opioids will dampen the full consequence of this plasticity. CGRP enhancement of synaptic transmission at the PB-CeLC synapse is through a postsynaptic mechanism. This mechanism could be through an increase in postsynaptic AMPARs. CGRP receptor antagonists also inhibit synaptic plasticity at the PB-CeLC synapse in brain slices from arthritic rats. Given this, endogenous CGRP could facilitate the synaptic plasticity produced by the nociceptive stimulus. Intra-CeLC injection of CGRP receptor antagonists before animals undergo the heat treatment could attenuate the synaptic plasticity and would be an important experiment to establish the role of CGRP in the induction of this synaptic plasticity.
As mentioned previously, exogenous application of opioids and CGRP have opposing effects on the PB-CeLC synapse and CeLC neurons, an interesting experiment would be to see what the endogenously released peptides do to this synapse and CeLC neurons. My lab has recently showed that a medium frequency synaptic stimulation can be used to release endogenous opioids (Winters et al., 2017). This protocol can be used to release opioids and CGRP to study their effects on the PB-CeLC synapse and CeLC neurons.
This thesis addressed the question of whether plasticity can outlast a stimulus using a novel approach. This plasticity was seen in the amygdala, an area important for associative learning and the affective component of pain. It has given a better understanding of the synaptic plasticity that occurs in pain conditions with no ongoing peripheral injury. It also showed that opioids and CGRP can modulate synapses important for pain in the amygdala.
References
ABRAHAM, W. C. 2008. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci, 9, 387.
ACOSTA, C. G. & LOPEZ, H. S. 1999. delta opioid receptor modulation of several voltage-dependent Ca(2+) currents in rat sensory neurons. J Neurosci, 19, 8337-48.
ADEDOYIN, M. O., VICINI, S. & NEALE, J. H. 2010. Endogenous N-acetylaspartylglutamate (NAAG) inhibits synaptic plasticity/transmission in the amygdala in a mouse inflammatory pain model. Mol Pain, 6, 60.
ADOLPHS, R., TRANEL, D., DAMASIO, H. & DAMASIO, A. 1994. Impaired recognition of emotion in facial expressions following bilateral damage to the human amygdala. Nature, 372, 669-72.
ALMEIDA, T. F., ROIZENBLATT, S. & TUFIK, S. 2004. Afferent pain pathways: a neuroanatomical review. Brain Res, 1000, 40-56.
ANSAH, O. B., BOURBIA, N., GONCALVES, L., ALMEIDA, A. & PERTOVAARA, A. 2010. Influence of amygdaloid glutamatergic receptors on sensory and emotional pain-related behavior in the neuropathic rat. Behav Brain Res, 209, 174-8.
ARRUDA-CARVALHO, M. & CLEM, R. L. 2015. Prefrontal-amygdala fear networks come into focus. Front Syst Neurosci, 9, 145.
ARVIDSSON, U., DADO, R. J., RIEDL, M., LEE, J. H., LAW, P. Y., LOH, H. H., ELDE, R. & WESSENDORF, M. W. 1995a. delta-Opioid receptor immunoreactivity: distribution in brainstem and spinal cord, and relationship to biogenic amines and enkephalin. J Neurosci, 15, 1215-35.
ARVIDSSON, U., RIEDL, M., CHAKRABARTI, S., LEE, J. H., NAKANO, A. H., DADO, R. J., LOH, H. H., LAW, P. Y., WESSENDORF, M. W. & ELDE, R. 1995b. Distribution and targeting of a mu-opioid receptor (MOR1) in brain and spinal cord. J Neurosci, 15, 3328-41.
ASCHAUER, D. F., KREUZ, S. & RUMPEL, S. 2013. Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS One, 8, e76310.
ASRAR, S., ZHOU, Z., REN, W. & JIA, Z. 2009. Ca(2+) permeable AMPA receptor induced long-term potentiation requires PI3/MAP kinases but not Ca/CaM-dependent kinase II. PLoS One, 4, e4339.
ATKINSON, J. H. 2004. Chronic back pain: searching for causes and cures. J Rheumatol, 31, 2323-5.
BALIKI, M. N., CHIALVO, D. R., GEHA, P. Y., LEVY, R. M., HARDEN, R. N., PARRISH, T. B. & APKARIAN, A. V. 2006. Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J Neurosci, 26, 12165-73.
BANKS, M. I., PEARCE, R. A. & SMITH, P. H. 1993. Hyperpolarization-activated cation current (Ih) in neurons of the medial nucleus of the trapezoid body: voltage-clamp analysis and enhancement by norepinephrine and cAMP suggest a modulatory mechanism in the auditory brain stem. J Neurophysiol, 70, 1420-32.
BASBAUM, A. I., BAUTISTA, D. M., SCHERRER, G. & JULIUS, D. 2009. Cellular and molecular mechanisms of pain. Cell, 139, 267-84.
BECHARA, A., TRANEL, D., DAMASIO, H., ADOLPHS, R., ROCKLAND, C. & DAMASIO, A. R. 1995. Double dissociation of conditioning and declarative knowledge relative to the amygdala and hippocampus in humans. Science, 269, 1115-8.
BERNARD, J. F., ALDEN, M. & BESSON, J. M. 1993. The organization of the efferent projections from the pontine parabrachial area to the amygdaloid complex: a Phaseolus vulgaris leucoagglutinin (PHA-L) study in the rat. J Comp Neurol, 329, 201-29.
BERNARD, J. F., HUANG, G. F. & BESSON, J. M. 1990. Effect of noxious somesthetic stimulation on the activity of neurons of the nucleus centralis of the amygdala. Brain Res, 523, 347-50.
BERNARD, J. F., HUANG, G. F. & BESSON, J. M. 1992. Nucleus centralis of the amygdala and the globus pallidus ventralis: electrophysiological evidence for an involvement in pain processes. J Neurophysiol, 68, 551-69.
BERNDT, A., SCHOENENBERGER, P., MATTIS, J., TYE, K. M., DEISSEROTH, K., HEGEMANN, P. & OERTNER, T. G. 2011. High-efficiency channelrhodopsins for fast neuronal stimulation at low light levels. Proc Natl Acad Sci U S A, 108, 7595-600.
BESTER, H., MATSUMOTO, N., BESSON, J. M. & BERNARD, J. F. 1997. Further evidence for the involvement of the spinoparabrachial pathway in nociceptive processes: a c-Fos study in the rat. J Comp Neurol, 383, 439-58.
BIEL, M., WAHL-SCHOTT, C., MICHALAKIS, S. & ZONG, X. 2009. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev, 89, 847-85.
BIRD, G. C., LASH, L. L., HAN, J. S., ZOU, X., WILLIS, W. D. & NEUGEBAUER, V. 2005. Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones. J Physiol, 564, 907-21.
BLAESSE, P., GOEDECKE, L., BAZELOT, M., CAPOGNA, M., PAPE, H. C. & JUNGLING, K. 2015. mu-Opioid Receptor-Mediated Inhibition of Intercalated Neurons and Effect on Synaptic Transmission to the Central Amygdala. J Neurosci, 35, 7317-25.
BLITS, B., DERKS, S., TWISK, J., EHLERT, E., PRINS, J. & VERHAAGEN, J. 2010. Adeno-associated viral vector (AAV)-mediated gene transfer in the red nucleus of the adult rat brain: comparative analysis of the transduction properties of seven AAV serotypes and lentiviral vectors. J Neurosci Methods, 185, 257-63.
BLYTH, F. M., MARCH, L. M., BRNABIC, A. J., JORM, L. R., WILLIAMSON, M. & COUSINS, M. J. 2001. Chronic pain in Australia: a prevalence study. Pain, 89, 127-34.
BORNHOVD, K., QUANTE, M., GLAUCHE, V., BROMM, B., WEILLER, C. & BUCHEL, C. 2002. Painful stimuli evoke different stimulus-response functions in the amygdala, prefrontal, insula and somatosensory cortex: a single-trial fMRI study. Brain, 125, 1326-36.
BORST, J. G. & SAKMANN, B. 1996. Calcium influx and transmitter release in a fast CNS synapse. Nature, 383, 431-4.
BORSZCZ, G. S. 1999. Differential contributions of medullary, thalamic and amygdaloid serotonin to the antinocieptive action of morphine administered into the periaqueductal gray: a model of morphine analgesia. Behavioral Neuroscience, 113, 612-631.
BOURGEAIS, L., GAURIAU, C. & BERNARD, J. F. 2001. Projections from the nociceptive area of the central nucleus of the amygdala to the forebrain: a PHA-L study in the rat. Eur J Neurosci, 14, 229-55.
BOYDEN, E. S., ZHANG, F., BAMBERG, E., NAGEL, G. & DEISSEROTH, K. 2005. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci, 8, 1263-8.
BUCHEL, C., BORNHOVD, K., QUANTE, M., GLAUCHE, V., BROMM, B. & WEILLER, C. 2002. Dissociable neural responses related to pain intensity, stimulus intensity, and stimulus awareness within the anterior cingulate cortex: a parametric single-trial laser functional magnetic resonance imaging study. J Neurosci, 22, 970-6.
BUSHNELL, M. C., CEKO, M. & LOW, L. A. 2013. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci, 14, 502-11.
BUSHNELL, M. C. & DUNCAN, G. H. 1989. Sensory and affective aspects of pain perception: is medial thalamus restricted to emotional issues? Exp Brain Res, 78, 415-8.
CAHILL, C. M., MCCLELLAN, K. A., MORINVILLE, A., HOFFERT, C., HUBATSCH, D., O’DONNELL, D. & BEAUDET, A. 2001a. Immunohistochemical distribution of delta opioid receptors in the rat central nervous system: evidence for somatodendritic labeling and antigen-specific cellular compartmentalization. J Comp Neurol, 440, 65-84.
CAHILL, C. M., MORINVILLE, A., HOFFERT, C., O’DONNELL, D. & BEAUDET, A. 2003. Up-regulation and trafficking of delta opioid receptor in a model of chronic inflammation: implications for pain control. Pain, 101, 199-208.
CAHILL, C. M., MORINVILLE, A., LEE, M. C., VINCENT, J. P., COLLIER, B. & BEAUDET, A. 2001b. Prolonged morphine treatment targets delta opioid receptors to neuronal plasma membranes and enhances delta-mediated antinociception. J Neurosci, 21, 7598-607.
CALVINO, B., LEVESQUE, G. & BESSON, J. M. 1982. Possible involvement of the amydaloid complex in morphine analgesia as studied by electrolytic lesions in rats. Brain Research, 233, 221-226.
CAMPEAU, S. & DAVIS, M. 1995. Involvement of the central nucleus and basolateral complex of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci, 15, 2301-11.
CANTERAS, N. S., SIMERLY, R. B. & SWANSON, L. W. 1994. Organization of projections from the ventromedial nucleus of the hypothalamus: a Phaseolus vulgaris-leucoagglutinin study in the rat. J Comp Neurol, 348, 41-79.
CARDINAL, R. N., PARKINSON, J. A., HALL, J. & EVERITT, B. J. 2002. Emotion and motivation: the role of the amygdala, ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev, 26, 321-52.
CARRASQUILLO, Y. & GEREAU, R. W. T. 2007. Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci, 27, 1543-51.
CARTER, M. E., SODEN, M. E., ZWEIFEL, L. S. & PALMITER, R. D. 2013. Genetic identification of a neural circuit that suppresses appetite. Nature, 503, 111-4.
CASTLE, M. J., PERLSON, E., HOLZBAUR, E. L. & WOLFE, J. H. 2014. Long-distance axonal transport of AAV9 is driven by dynein and kinesin-2 and is trafficked in a highly motile Rab7-positive compartment. Mol Ther, 22, 554-66.
CEARLEY, C. N. & WOLFE, J. H. 2007. A single injection of an adeno-associated virus vector into nuclei with divergent connections results in widespread vector distribution in the brain and global correction of a neurogenetic disease. J Neurosci, 27, 9928-40.
CHAMBERLIN, N. L., MANSOUR, A., WATSON, S. J. & SAPER, C. B. 1999. Localization of mu-opioid receptors on amygdaloid projection neurons in the parabrachial nucleus of the rat. Brain Res, 827, 198-204.
CHAPLAN, S. R., BACH, F. W., POGREL, J. W., CHUNG, J. M. & YAKSH, T. L. 1994. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods, 53, 55-63.
CHATER, T. E. & GODA, Y. 2014. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front Cell Neurosci, 8, 401.
CHEN, T., KOGA, K., DESCALZI, G., QIU, S., WANG, J., ZHANG, L. S., ZHANG, Z. J., HE, X. B., QIN, X., XU, F. Q., HU, J., WEI, F., HUGANIR, R. L., LI, Y. Q. & ZHUO, M. 2014. Postsynaptic potentiation of corticospinal projecting neurons in the anterior cingulate cortex after nerve injury. Mol Pain, 10, 33.
CHENG, P. Y., LIU-CHEN, L. Y. & PICKEL, V. M. 1997. Dual ultrastructural immunocytochemical labeling of mu and delta opioid receptors in the superficial layers of the rat cervical spinal cord. Brain Res, 778, 367-80.
CHENG, S. J., CHEN, C. C., YANG, H. W., CHANG, Y. T., BAI, S. W., CHEN, C. C., YEN, C. T. & MIN, M. Y. 2011. Role of extracellular signal-regulated kinase in synaptic transmission and plasticity of a nociceptive input on capsular central amygdaloid neurons in normal and acid-induced muscle pain mice. J Neurosci, 31, 2258-70.
CHIENG, B. & CHRISTIE, M. J. 1994. Hyperpolarization by opioids acting on mu-receptors of a sub-population of rat periaqueductal gray neurones in vitro. Br J Pharmacol, 113, 121-8.
CHIENG, B. & CHRISTIE, M. J. 2009. Chronic morphine treatment induces functional delta-opioid receptors in amygdala neurons that project to periaqueductal grey. Neuropharmacology, 57, 430-7.
CHIENG, B. & WILLIAMS, J. T. 1998. Increased opioid inhibition of GABA release in nucleus accumbens during morphine withdrawal. J Neurosci, 18, 7033-9.
CHIENG, B. C., CHRISTIE, M. J. & OSBORNE, P. B. 2006. Characterization of neurons in the rat central nucleus of the amygdala: cellular physiology, morphology, and opioid sensitivity. J Comp Neurol, 497, 910-27.
CHOW, B. Y., HAN, X., DOBRY, A. S., QIAN, X., CHUONG, A. S., LI, M., HENNINGER, M. A., BELFORT, G. M., LIN, Y., MONAHAN, P. E. & BOYDEN, E. S. 2010. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature, 463, 98-102.
CHRISTIE, M. J. & NORTH, R. A. 1988. Agonists at mu-opioid, M2-muscarinic and GABAB-receptors increase the same potassium conductance in rat lateral parabrachial neurones. Br J Pharmacol, 95, 896-902.
CHU, H. Y. & ZHEN, X. 2010. Hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels in the regulation of midbrain dopamine systems. Acta Pharmacol Sin, 31, 1036-43.
CIOCCHI, S., HERRY, C., GRENIER, F., WOLFF, S. B., LETZKUS, J. J., VLACHOS, I., EHRLICH, I., SPRENGEL, R., DEISSEROTH, K., STADLER, M. B., MULLER, C. & LUTHI, A. 2010. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature, 468, 277-82.
CITRI, A. & MALENKA, R. C. 2008. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology, 33, 18-41.
CODERRE, T. J., KATZ, J., VACCARINO, A. L. & MELZACK, R. 1993. Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain, 52, 259-85.
COLLIER, H. O. & ROY, A. C. 1974. Morphine-like drugs inhibit the stimulation of E prostaglandins of cyclic AMP formation by rat brain homogenate. Nature, 248, 24-7.
COLLINGRIDGE, G. L., ISAAC, J. T. & WANG, Y. T. 2004. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci, 5, 952-62.
CONNOR, M. & CHRISTIE, M. D. 1999. Opioid receptor signalling mechanisms. Clin Exp Pharmacol Physiol, 26, 493-9.
CONNOR, M., SCHULLER, A., PINTAR, J. E. & CHRISTIE, M. J. 1999. Mu-opioid receptor modulation of calcium channel current in periaqueductal grey neurons from C57B16/J mice and mutant mice lacking MOR-1. Br J Pharmacol, 126, 1553-8.
COOK, A. J., WOOLF, C. J., WALL, P. D. & MCMAHON, S. B. 1987. Dynamic receptive field plasticity in rat spinal cord dorsal horn following C-primary afferent input. Nature, 325, 151-3.
COOPER, D. M., LONDOS, C., GILL, D. L. & RODBELL, M. 1982. Opiate receptor-mediated inhibition of adenylate cyclase in rat striatal plasma membranes. J Neurochem, 38, 1164-7.
CRICK, F. H. 1979. Thinking about the brain. Sci Am, 241, 219-32.
CRUIKSHANK, S. J., URABE, H., NURMIKKO, A. V. & CONNORS, B. W. 2010. Pathway-specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron, 65, 230-45.
DAHL, J. B., BRENNUM, J., ARENDT-NIELSEN, L., JENSEN, T. S. & KEHLET, H. 1993. The effect of pre- versus postinjury infiltration with lidocaine on thermal and mechanical hyperalgesia after heat injury to the skin. Pain, 53, 43-51.
DEISSEROTH, K. 2011. Optogenetics. Nat Methods, 8, 26-9.
DELANEY, A. J., CRANE, J. W. & SAH, P. 2007. Noradrenaline modulates transmission at a central synapse by a presynaptic mechanism. Neuron, 56, 880-92.
DI PASQUALE, G., DAVIDSON, B. L., STEIN, C. S., MARTINS, I., SCUDIERO, D., MONKS, A. & CHIORINI, J. A. 2003. Identification of PDGFR as a receptor for AAV-5 transduction. Nat Med, 9, 1306-12.
DICKENSON, A. H. 1991. Mechanisms of the analgesic actions of opiates and opioids. Br Med Bull, 47, 690-702.
DIFRANCESCO, D. & TORTORA, P. 1991. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature, 351, 145-7.
DING, Y. Q., KANEKO, T., NOMURA, S. & MIZUNO, N. 1996. Immunohistochemical localization of mu-opioid receptors in the central nervous system of the rat. J Comp Neurol, 367, 375-402.
DISA, J., PARAMESWARAN, N., NAMBI, P. & AIYAR, N. 2000. Involvement of cAMP-dependent protein kinase and pertussis toxin-sensitive G-proteins in CGRP mediated JNK activation in human neuroblastoma cell line. Neuropeptides, 34, 229-33.
DONG, Y. L., FUKAZAWA, Y., WANG, W., KAMASAWA, N. & SHIGEMOTO, R. 2010. Differential postsynaptic compartments in the laterocapsular division of the central nucleus of amygdala for afferents from the parabrachial nucleus and the basolateral nucleus in the rat. J Comp Neurol, 518, 4771-91.
DOODS, H., ARNDT, K., RUDOLF, K. & JUST, S. 2007. CGRP antagonists: unravelling the role of CGRP in migraine. Trends Pharmacol Sci, 28, 580-7.
DORON, N. N. & LEDOUX, J. E. 1999. Organization of projections to the lateral amygdala from auditory and visual areas of the thalamus in the rat. J Comp Neurol, 412, 383-409.
DRISSI, H., LASMOLES, F., LE MELLAY, V., MARIE, P. J. & LIEBERHERR, M. 1998. Activation of phospholipase C-beta1 via Galphaq/11 during calcium mobilization by calcitonin gene-related peptide. J Biol Chem, 273, 20168-74.
ELMAN, I. & BORSOOK, D. 2016. Common Brain Mechanisms of Chronic Pain and Addiction. Neuron, 89, 11-36.
ERBS, E., FAGET, L., SCHERRER, G., MATIFAS, A., FILLIOL, D., VONESCH, J. L., KOCH, M., KESSLER, P., HENTSCH, D., BIRLING, M. C., KOUTSOURAKIS, M., VASSEUR, L., VEINANTE, P., KIEFFER, B. L. & MASSOTTE, D. 2015. A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct Funct, 220, 677-702.
EVANS, B. N., ROSENBLATT, M. I., MNAYER, L. O., OLIVER, K. R. & DICKERSON, I. M. 2000. CGRP-RCP, a novel protein required for signal transduction at calcitonin gene-related peptide and adrenomedullin receptors. J Biol Chem, 275, 31438-43.
FABER, E. S. & SAH, P. 2004. Opioids inhibit lateral amygdala pyramidal neurons by enhancing a dendritic potassium current. J Neurosci, 24, 3031-9.
FERRARI, F. K., SAMULSKI, T., SHENK, T. & SAMULSKI, R. J. 1996. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol, 70, 3227-34.
FINNEGAN, T. F., CHEN, S. R. & PAN, H. L. 2005. Effect of the {mu} opioid on excitatory and inhibitory synaptic inputs to periaqueductal gray-projecting neurons in the amygdala. J Pharmacol Exp Ther, 312, 441-8.
FOLTZ, E. L. & WHITE, L. E., JR. 1962. Pain “relief” by frontal cingulumotomy. J Neurosurg, 19, 89-100.
FRASER, G. L., GAUDREAU, G. A., CLARKE, P. B., MENARD, D. P. & PERKINS, M. N. 2000. Antihyperalgesic effects of delta opioid agonists in a rat model of chronic inflammation. Br J Pharmacol, 129, 1668-72.
FU, Y., HAN, J., ISHOLA, T., SCERBO, M., ADWANIKAR, H., RAMSEY, C. & NEUGEBAUER, V. 2008. PKA and ERK, but not PKC, in the amygdala contribute to pain-related synaptic plasticity and behavior. Mol Pain, 4, 26.
FU, Y. & NEUGEBAUER, V. 2008. Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain-related synaptic facilitation and behavior. J Neurosci, 28, 3861-76.
FUNADA, M., SUZUKI, T., NARITA, M., MISAWA, M. & NAGASE, H. 1993. Blockade of morphine reward through the activation of kappa-opioid receptors in mice. Neuropharmacology, 32, 1315-23.
GAO, Y. J. & JI, R. R. 2009. c-Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? Open Pain J, 2, 11-17.
GAO, Y. J., REN, W. H., ZHANG, Y. Q. & ZHAO, Z. Q. 2004. Contributions of the anterior cingulate cortex and amygdala to pain- and fear-conditioned place avoidance in rats. Pain, 110, 343-53.
GINGOLD, S. I., GREENSPAN, J. D. & APKARIAN, A. V. 1991. Anatomic evidence of nociceptive inputs to primary somatosensory cortex: relationship between spinothalamic terminals and thalamocortical cells in squirrel monkeys. J Comp Neurol, 308, 467-90.
GOOD, C. H., WANG, H., CHEN, Y. H., MEJIAS-APONTE, C. A., HOFFMAN, A. F. & LUPICA, C. R. 2013. Dopamine D4 receptor excitation of lateral habenula neurons via multiple cellular mechanisms. J Neurosci, 33, 16853-64.
GRADINARU, V., THOMPSON, K. R. & DEISSEROTH, K. 2008. eNpHR: a Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol, 36, 129-39.
GREGOIRE, S., WATTIEZ, A. S., ETIENNE, M., MARCHAND, F. & ARDID, D. 2014. Monoarthritis-induced emotional and cognitive impairments in rats are sensitive to low systemic doses or intra-amygdala injections of morphine. Eur J Pharmacol, 735, 1-9.
GUILBAUD, G., PESCHANSKI, M., GAUTRON, M. & BINDER, D. 1980. Neurones responding to noxious stimulation in VB complex and caudal adjacent regions in the thalamus of the rat. Pain, 8, 303-18.
GUIRE, E. S., OH, M. C., SODERLING, T. R. & DERKACH, V. A. 2008. Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J Neurosci, 28, 6000-9.
GUNAYDIN, L. A., YIZHAR, O., BERNDT, A., SOHAL, V. S., DEISSEROTH, K. & HEGEMANN, P. 2010. Ultrafast optogenetic control. Nat Neurosci, 13, 387-92.
GUREJE, O., VON KORFF, M., SIMON, G. E. & GATER, R. 1998. Persistent pain and well-being: a World Health Organization Study in Primary Care. JAMA, 280, 147-51.
HACK, S. P., BAGLEY, E. E., CHIENG, B. C. & CHRISTIE, M. J. 2005. Induction of delta-opioid receptor function in the midbrain after chronic morphine treatment. J Neurosci, 25, 3192-8.
HAN, J. S., ADWANIKAR, H., LI, Z., JI, G. & NEUGEBAUER, V. 2010. Facilitation of synaptic transmission and pain responses by CGRP in the amygdala of normal rats. Mol Pain, 6, 10.
HAN, J. S., LI, W. & NEUGEBAUER, V. 2005. Critical role of calcitonin gene-related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J Neurosci, 25, 10717-28.
HAN, J. S. & NEUGEBAUER, V. 2004. Synaptic plasticity in the amygdala in a visceral pain model in rats. Neurosci Lett, 361, 254-7.
HAN, J. S. & NEUGEBAUER, V. 2005. mGluR1 and mGluR5 antagonists in the amygdala inhibit different components of audible and ultrasonic vocalizations in a model of arthritic pain. Pain, 113, 211-22.
HAN, S., SOLEIMAN, M. T., SODEN, M. E., ZWEIFEL, L. S. & PALMITER, R. D. 2015. Elucidating an Affective Pain Circuit that Creates a Threat Memory. Cell, 162, 363-74.
HARGREAVES, K., DUBNER, R., BROWN, F., FLORES, C. & JORIS, J. 1988. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain, 32, 77-88.
HARING, C., HUMPEL, C., SKOFITSCH, G., KROBATH, J., JAVORSKY, F. & SARIA, A. 1991. Calcitonin gene-related peptide in the amygdaloid complex of the rat: immunohistochemical and quantitative distribution, and drug effects on calcium dependent, potassium-evoked in vitro release. Synapse, 8, 261-9.
HAUBENSAK, W., KUNWAR, P. S., CAI, H., CIOCCHI, S., WALL, N. R., PONNUSAMY, R., BIAG, J., DONG, H. W., DEISSEROTH, K., CALLAWAY, E. M., FANSELOW, M. S., LUTHI, A. & ANDERSON, D. J. 2010. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature, 468, 270-6.
HAY, D. L., CHRISTOPOULOS, G., CHRISTOPOULOS, A. & SEXTON, P. M. 2006. Determinants of 1-piperidinecarboxamide, N-[2-[[5-amino-l-[[4-(4-pyridinyl)-l-piperazinyl]carbonyl]pentyl]amino]-1-[(3,5-d ibromo-4-hydroxyphenyl)methyl]-2-oxoethyl]-4-(1,4-dihydro-2-oxo-3(2H)-quinazoliny l) (BIBN4096BS) affinity for calcitonin gene-related peptide and amylin receptors–the role of receptor activity modifying protein 1. Mol Pharmacol, 70, 1984-91.
HAY, D. L., HOWITT, S. G., CONNER, A. C., SCHINDLER, M., SMITH, D. M. & POYNER, D. R. 2003. CL/RAMP2 and CL/RAMP3 produce pharmacologically distinct adrenomedullin receptors: a comparison of effects of adrenomedullin22-52, CGRP8-37 and BIBN4096BS. Br J Pharmacol, 140, 477-86.
HEBERT, M. A., ARDID, D., HENRIE, J. A., TAMASHIRO, K., BLANCHARD, D. C. & BLANCHARD, R. J. 1999. Amygdala lesions produce analgesia in a novel, ethologically relevant acute pain test. Physiol Behav, 67, 99-105.
HEINRICHER, M. M., TAVARES, I., LEITH, J. L. & LUMB, B. M. 2009. Descending control of nociception: Specificity, recruitment and plasticity. Brain Res Rev, 60, 214-25.
HELMSTETTER, F. J. & BELLGOWAN, P. S. 1994. Effects of muscimol applied to the basolateral amygdala on acquisition and expression of contextual fear conditioning in rats. Behav Neurosci, 108, 1005-9.
HELMSTETTER, F. J. & FANSELOW, M. S. 1987. Effects of naltrexone on learning and performance of conditional fear-induced freezing and opioid analgesia. Physiol Behav, 39, 501-5.
HELMSTETTER, F. J. & LANDEIRA-FERNANDEZ, J. 1990. Conditional hypoalgesia is attenuated by naltrexone applied to the periaqueductal gray. Brain Res, 537, 88-92.
HITCHCOCK, J. & DAVIS, M. 1986. Lesions of the amygdala, but not of the cerebellum or red nucleus, block conditioned fear as measured with the potentiated startle paradigm. Behav Neurosci, 100, 11-22.
HOLDRIDGE, S. V. & CAHILL, C. M. 2007. Spinal administration of a delta opioid receptor agonist attenuates hyperalgesia and allodynia in a rat model of neuropathic pain. Eur J Pain, 11, 685-93.
HUANG, C. L., SLESINGER, P. A., CASEY, P. J., JAN, Y. N. & JAN, L. Y. 1995. Evidence that direct binding of G beta gamma to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron, 15, 1133-43.
HUANG, G. F., BESSON, J. M. & BERNARD, J. F. 1993a. Intravenous morphine depresses the transmission of noxious messages to the nucleus centralis of the amygdala. Eur J Pharmacol, 236, 449-56.
HUANG, G. F., BESSON, J. M. & BERNARD, J. F. 1993b. Morphine depresses the transmission of noxious messages in the spino(trigemino)-ponto-amygdaloid pathway. Eur J Pharmacol, 230, 279-84.
IKEDA, R., TAKAHASHI, Y., INOUE, K. & KATO, F. 2007. NMDA receptor-independent synaptic plasticity in the central amygdala in the rat model of neuropathic pain. Pain, 127, 161-72.
INGRAM, S. L., VAUGHAN, C. W., BAGLEY, E. E., CONNOR, M. & CHRISTIE, M. J. 1998. Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci, 18, 10269-76.
INGRAM, S. L. & WILLIAMS, J. T. 1994. Opioid inhibition of Ih via adenylyl cyclase. Neuron, 13, 179-86.
INTURRISI, C. E. 2002. Clinical pharmacology of opioids for pain. Clin J Pain, 18, S3-13.
ISAAC, J. T., ASHBY, M. C. & MCBAIN, C. J. 2007. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron, 54, 859-71.
JACKMAN, S. L., BENEDUCE, B. M., DREW, I. R. & REGEHR, W. G. 2014. Achieving high-frequency optical control of synaptic transmission. J Neurosci, 34, 7704-14.
JI, G., FU, Y., ADWANIKAR, H. & NEUGEBAUER, V. 2013. Non-pain-related CRF1 activation in the amygdala facilitates synaptic transmission and pain responses. Mol Pain, 9, 2.
JI, G., SUN, H., FU, Y., LI, Z., PAIS-VIEIRA, M., GALHARDO, V. & NEUGEBAUER, V. 2010. Cognitive impairment in pain through amygdala-driven prefrontal cortical deactivation. J Neurosci, 30, 5451-64.
JI, R. R., KOHNO, T., MOORE, K. A. & WOOLF, C. J. 2003. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci, 26, 696-705.
JOHANSEN, J. P., FIELDS, H. L. & MANNING, B. H. 2001. The affective component of pain in rodents: direct evidence for a contribution of the anterior cingulate cortex. Proc Natl Acad Sci U S A, 98, 8077-82.
JOLKKONEN, E. & PITKANEN, A. 1998. Intrinsic connections of the rat amygdaloid complex: projections originating in the central nucleus. J Comp Neurol, 395, 53-72.
KANDEL, E. R. 2013. Principles of neural science, New York, McGraw-Hill.
KATZ, B. & MILEDI, R. 1967a. A study of synaptic transmission in the absence of nerve impulses. J Physiol, 192, 407-36.
KATZ, B. & MILEDI, R. 1967b. The timing of calcium action during neuromuscular transmission. J Physiol, 189, 535-44.
KAUER, J. A. & MALENKA, R. C. 2007. Synaptic plasticity and addiction. Nat Rev Neurosci, 8, 844-58.
KENSHALO, D. R., IWATA, K., SHOLAS, M. & THOMAS, D. A. 2000. Response properties and organization of nociceptive neurons in area 1 of monkey primary somatosensory cortex. J Neurophysiol, 84, 719-29.
KENSHALO, D. R., JR., CHUDLER, E. H., ANTON, F. & DUBNER, R. 1988. SI nociceptive neurons participate in the encoding process by which monkeys perceive the intensity of noxious thermal stimulation. Brain Res, 454, 378-82.
KIEFFER, B. L. 1999. Opioids: first lessons from knockout mice. Trends Pharmacol Sci, 20, 19-26.
KLUVER, H. & BUCY, P. C. 1997. Preliminary analysis of functions of the temporal lobes in monkeys. 1939. J Neuropsychiatry Clin Neurosci, 9, 606-20.
KNOLL, A. T., MUSCHAMP, J. W., SILLIVAN, S. E., FERGUSON, D., DIETZ, D. M., MELONI, E. G., CARROLL, F. I., NESTLER, E. J., KONRADI, C. & CARLEZON, W. A., JR. 2011. Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biol Psychiatry, 70, 425-33.
KOLBER, B. J., MONTANA, M. C., CARRASQUILLO, Y., XU, J., HEINEMANN, S. F., MUGLIA, L. J. & GEREAU, R. W. T. 2010. Activation of metabotropic glutamate receptor 5 in the amygdala modulates pain-like behavior. J Neurosci, 30, 8203-13.
KRUGER, L., STERNINI, C., BRECHA, N. C. & MANTYH, P. W. 1988. Distribution of calcitonin gene-related peptide immunoreactivity in relation to the rat central somatosensory projection. J Comp Neurol, 273, 149-62.
KUNER, R. 2010. Central mechanisms of pathological pain. Nat Med, 16, 1258-66.
KUPERS, R. C., KONINGS, H., ADRIAENSEN, H. & GYBELS, J. M. 1991. Morphine differentially affects the sensory and affective pain ratings in neurogenic and idiopathic forms of pain. Pain, 47, 5-12.
KWAK, J. 2012. Capsaicin Blocks the Hyperpolarization-Activated Inward Currents via TRPV1 in the Rat Dorsal Root Ganglion Neurons. Exp Neurobiol, 21, 75-82.
LAGRAIZE, S. C., BORZAN, J., PENG, Y. B. & FUCHS, P. N. 2006. Selective regulation of pain affect following activation of the opioid anterior cingulate cortex system. Exp Neurol, 197, 22-30.
LAND, B. B., BRUCHAS, M. R., LEMOS, J. C., XU, M., MELIEF, E. J. & CHAVKIN, C. 2008. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci, 28, 407-14.
LANYI, J. K. & OESTERHELT, D. 1982. Identification of the retinal-binding protein in halorhodopsin. J Biol Chem, 257, 2674-7.
LATREMOLIERE, A. & WOOLF, C. J. 2009. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain, 10, 895-926.
LAU, B. K. & VAUGHAN, C. W. 2014. Descending modulation of pain: the GABA disinhibition hypothesis of analgesia. Curr Opin Neurobiol, 29, 159-64.
LE MERRER, J., BECKER, J. A., BEFORT, K. & KIEFFER, B. L. 2009. Reward processing by the opioid system in the brain. Physiol Rev, 89, 1379-412.
LEDOUX, J. E. 2000. Emotion circuits in the brain. Annu Rev Neurosci, 23, 155-84.
LEDOUX, J. E., CICCHETTI, P., XAGORARIS, A. & ROMANSKI, L. M. 1990a. The lateral amygdaloid nucleus: sensory interface of the amygdala in fear conditioning. J Neurosci, 10, 1062-9.
LEDOUX, J. E., FARB, C. & RUGGIERO, D. A. 1990b. Topographic organization of neurons in the acoustic thalamus that project to the amygdala. J Neurosci, 10, 1043-54.
LEDOUX, J. E., IWATA, J., CICCHETTI, P. & REIS, D. J. 1988. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci, 8, 2517-29.
LI, H., PENZO, M. A., TANIGUCHI, H., KOPEC, C. D., HUANG, Z. J. & LI, B. 2013. Experience-dependent modification of a central amygdala fear circuit. Nat Neurosci, 16, 332-9.
LI, Z., JI, G. & NEUGEBAUER, V. 2011. Mitochondrial reactive oxygen species are activated by mGluR5 through IP3 and activate ERK and PKA to increase excitability of amygdala neurons and pain behavior. J Neurosci, 31, 1114-27.
LIN, J. Y. 2011. A user’s guide to channelrhodopsin variants: features, limitations and future developments. Exp Physiol, 96, 19-25.
LOH, H. H., LIU, H. C., CAVALLI, A., YANG, W., CHEN, Y. F. & WEI, L. N. 1998. mu Opioid receptor knockout in mice: effects on ligand-induced analgesia and morphine lethality. Brain Res Mol Brain Res, 54, 321-6.
LU, Y. C., CHEN, Y. Z., WEI, Y. Y., HE, X. T., LI, X., HU, W., YANAGAWA, Y., WANG, W., WU, S. X. & DONG, Y. L. 2015. Neurochemical properties of the synapses between the parabrachial nucleus-derived CGRP-positive axonal terminals and the GABAergic neurons in the lateral capsular division of central nucleus of amygdala. Mol Neurobiol, 51, 105-18.
LUO, C., KUNER, T. & KUNER, R. 2014. Synaptic plasticity in pathological pain. Trends Neurosci, 37, 343-55.
LUSCHER, C. & SLESINGER, P. A. 2010. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci, 11, 301-15.
MALENKA, R. C. & NICOLL, R. A. 1999. Long-term potentiation–a decade of progress? Science, 285, 1870-4.
MANNING, B. H. & MEYER, D. J. 1995. The central nucleus of the amygdala contributes to the production of morphine antinociception in the rat tail-flick test. The Journal of Neuroscience, 15, 8199-8213.
MANSOUR, A., FOX, C. A., AKIL, H. & WATSON, S. J. 1995. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci, 18, 22-9.
MANSOUR, A., FOX, C. A., BURKE, S., MENG, F., THOMPSON, R. C., AKIL, H. & WATSON, S. J. 1994. Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J Comp Neurol, 350, 412-38.
MAREK, R., STROBEL, C., BREDY, T. W. & SAH, P. 2013. The amygdala and medial prefrontal cortex: partners in the fear circuit. J Physiol, 591, 2381-91.
MARTIN, W. R. 1979. History and development of mixed opioid agonists, partial agonists and antagonists. Br J Clin Pharmacol, 7 Suppl 3, 273s-279s.
MATSUNO-YAGI, A. & MUKOHATA, Y. 1977. Two possible roles of bacteriorhodopsin; a comparative study of strains of Halobacterium halobium differing in pigmentation. Biochem Biophys Res Commun, 78, 237-43.
MATTHES, H. W., MALDONADO, R., SIMONIN, F., VALVERDE, O., SLOWE, S., KITCHEN, I., BEFORT, K., DIERICH, A., LE MEUR, M., DOLLE, P., TZAVARA, E., HANOUNE, J., ROQUES, B. P. & KIEFFER, B. L. 1996. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature, 383, 819-23.
MATTHES, H. W., SMADJA, C., VALVERDE, O., VONESCH, J. L., FOUTZ, A. S., BOUDINOT, E., DENAVIT-SAUBIE, M., SEVERINI, C., NEGRI, L., ROQUES, B. P., MALDONADO, R. & KIEFFER, B. L. 1998. Activity of the delta-opioid receptor is partially reduced, whereas activity of the kappa-receptor is maintained in mice lacking the mu-receptor. J Neurosci, 18, 7285-95.
MATTIS, J., TYE, K. M., FERENCZI, E. A., RAMAKRISHNAN, C., O’SHEA, D. J., PRAKASH, R., GUNAYDIN, L. A., HYUN, M., FENNO, L. E., GRADINARU, V., YIZHAR, O. & DEISSEROTH, K. 2011. Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat Methods, 9, 159-72.
MCCARTY, D. M., MONAHAN, P. E. & SAMULSKI, R. J. 2001. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther, 8, 1248-54.
MCCORMICK, D. A. & PAPE, H. C. 1990. Noradrenergic and serotonergic modulation of a hyperpolarization-activated cation current in thalamic relay neurones. J Physiol, 431, 319-42.
MCDONALD, A. J. 1982. Cytoarchitecture of the central amygdaloid nucleus of the rat. J Comp Neurol, 208, 401-18.
MCDONALD, A. J. & MASCAGNI, F. 1997. Projections of the lateral entorhinal cortex to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience, 77, 445-59.
MCDONALD, A. J., MASCAGNI, F. & GUO, L. 1996. Projections of the medial and lateral prefrontal cortices to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience, 71, 55-75.
MCLATCHIE, L. M., FRASER, N. J., MAIN, M. J., WISE, A., BROWN, J., THOMPSON, N., SOLARI, R., LEE, M. G. & FOORD, S. M. 1998. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature, 393, 333-9.
MCLAUGHLIN, J. P., MARTON-POPOVICI, M. & CHAVKIN, C. 2003. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci, 23, 5674-83.
MIKA, J., PRZEWLOCKI, R. & PRZEWLOCKA, B. 2001. The role of delta-opioid receptor subtypes in neuropathic pain. Eur J Pharmacol, 415, 31-7.
MILLAN, M. J. 2002. Descending control of pain. Prog Neurobiol, 66, 355-474.
MOGA, M. M., WEIS, R. P. & MOORE, R. Y. 1995. Efferent projections of the paraventricular thalamic nucleus in the rat. J Comp Neurol, 359, 221-38.
MOISES, H. C., RUSIN, K. I. & MACDONALD, R. L. 1994. mu-Opioid receptor-mediated reduction of neuronal calcium current occurs via a G(o)-type GTP-binding protein. J Neurosci, 14, 3842-51.
MORITA, D., RAH, J. C. & ISAAC, J. T. 2014. Incorporation of inwardly rectifying AMPA receptors at silent synapses during hippocampal long-term potentiation. Philos Trans R Soc Lond B Biol Sci, 369, 20130156.
MUCHA, R. F. & HERZ, A. 1985. Motivational properties of kappa and mu opioid receptor agonists studied with place and taste preference conditioning. Psychopharmacology (Berl), 86, 274-80.
NAGEL, G., BRAUNER, M., LIEWALD, J. F., ADEISHVILI, N., BAMBERG, E. & GOTTSCHALK, A. 2005. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol, 15, 2279-84.
NAGEL, G., OLLIG, D., FUHRMANN, M., KATERIYA, S., MUSTI, A. M., BAMBERG, E. & HEGEMANN, P. 2002. Channelrhodopsin-1: a light-gated proton channel in green algae. Science, 296, 2395-8.
NAGEL, G., SZELLAS, T., HUHN, W., KATERIYA, S., ADEISHVILI, N., BERTHOLD, P., OLLIG, D., HEGEMANN, P. & BAMBERG, E. 2003. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A, 100, 13940-5.
NATURA, G., VON BANCHET, G. S. & SCHAIBLE, H. G. 2005. Calcitonin gene-related peptide enhances TTX-resistant sodium currents in cultured dorsal root ganglion neurons from adult rats. Pain, 116, 194-204.
NEUGEBAUER, V. 2015. Amygdala pain mechanisms. Handb Exp Pharmacol, 227, 261-84.
NEUGEBAUER, V. & LI, W. 2002. Processing of nociceptive mechanical and thermal information in central amygdala neurons with knee-joint input. J Neurophysiol, 87, 103-12.
NEUGEBAUER, V. & LI, W. 2003. Differential sensitization of amygdala neurons to afferent inputs in a model of arthritic pain. J Neurophysiol, 89, 716-27.
NEUGEBAUER, V., LI, W., BIRD, G. C., BHAVE, G. & GEREAU, R. W. T. 2003. Synaptic plasticity in the amygdala in a model of arthritic pain: differential roles of metabotropic glutamate receptors 1 and 5. J Neurosci, 23, 52-63.
NEUGEBAUER, V., RUMENAPP, P. & SCHAIBLE, H. G. 1996. Calcitonin gene-related peptide is involved in the spinal processing of mechanosensory input from the rat’s knee joint and in the generation and maintenance of hyperexcitability of dorsal horn-neurons during development of acute inflammation. Neuroscience, 71, 1095-109.
NIKITENKO, L. L., BLUCHER, N., FOX, S. B., BICKNELL, R., SMITH, D. M. & REES, M. C. 2006. Adrenomedullin and CGRP interact with endogenous calcitonin-receptor-like receptor in endothelial cells and induce its desensitisation by different mechanisms. J Cell Sci, 119, 910-22.
NOTOMI, T. & SHIGEMOTO, R. 2004. Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J Comp Neurol, 471, 241-76.
OESTERHELT, D. & STOECKENIUS, W. 1971. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nat New Biol, 233, 149-52.
OESTERHELT, D. & STOECKENIUS, W. 1973. Functions of a new photoreceptor membrane. Proc Natl Acad Sci U S A, 70, 2853-7.
OLDHAM, W. M. & HAMM, H. E. 2008. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol, 9, 60-71.
OLIVER, K. R., WAINWRIGHT, A., HEAVENS, R. P., HILL, R. G. & SIRINATHSINGHJI, D. J. 1998. Distribution of novel CGRP1 receptor and adrenomedullin receptor mRNAs in the rat central nervous system. Brain Res Mol Brain Res, 57, 149-54.
OLIVERAS, J. L., MAIXNER, W., DUBNER, R., BUSHNELL, M. C., DUNCAN, G., THOMAS, D. A. & BATES, R. 1986. Dorsal horn opiate administration attenuates the perceived intensity of noxious heat stimulation in behaving monkey. Brain Res, 371, 368-71.
ORTINSKI, P. I., DONG, J., MUNGENAST, A., YUE, C., TAKANO, H., WATSON, D. J., HAYDON, P. G. & COULTER, D. A. 2010. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci, 13, 584-91.
PAPE, H. C. & PARE, D. 2010. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev, 90, 419-63.
PAXINOS, G. & WATSON, C. 1986. The rat brain in stereotaxic coordinates, Sydney, Academic Press.
PEDERSEN, L. H., SCHEEL-KRUGER, J. & BLACKBURN-MUNRO, G. 2007. Amygdala GABA-A receptor involvement in mediating sensory-discriminative and affective-motivational pain responses in a rat model of peripheral nerve injury. Pain, 127, 17-26.
PESCHANSKI, M., GUILBAUD, G., GAUTRON, M. & BESSON, J. M. 1980. Encoding of noxious heat messages in neurons of the ventrobasal thalamic complex of the rat. Brain Res, 197, 401-13.
PETRILLO, P., ANGELICI, O., BINGHAM, S., FICALORA, G., GARNIER, M., ZARATIN, P. F., PETRONE, G., POZZI, O., SBACCHI, M., STEAN, T. O., UPTON, N., DONDIO, G. M. & SCHEIDELER, M. A. 2003. Evidence for a selective role of the delta-opioid agonist [8R-(4bS*,8aalpha,8abeta, 12bbeta)]7,10-Dimethyl-1-methoxy-11-(2-methylpropyl)oxycarbonyl 5,6,7,8,12,12b-hexahydro-(9H)-4,8-methanobenzofuro[3,2-e]pyrrolo[2,3-g]isoquinoli ne hydrochloride (SB-235863) in blocking hyperalgesia associated with inflammatory and neuropathic pain responses. J Pharmacol Exp Ther, 307, 1079-89.
PFEIFFER, A., BRANTL, V., HERZ, A. & EMRICH, H. M. 1986. Psychotomimesis mediated by kappa opiate receptors. Science, 233, 774-6.
PHILLIPS, C. J. 2009. The Cost and Burden of Chronic Pain. Rev Pain, 3, 2-5.
PITKANEN, A., SAVANDER, V. & LEDOUX, J. E. 1997. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci, 20, 517-23.
PLANT, K., PELKEY, K. A., BORTOLOTTO, Z. A., MORITA, D., TERASHIMA, A., MCBAIN, C. J., COLLINGRIDGE, G. L. & ISAAC, J. T. 2006. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci, 9, 602-4.
POULIN, J. F., CHEVALIER, B., LAFOREST, S. & DROLET, G. 2006. Enkephalinergic afferents of the centromedial amygdala in the rat. J Comp Neurol, 496, 859-76.
PRADHAN, A. A., BEFORT, K., NOZAKI, C., GAVERIAUX-RUFF, C. & KIEFFER, B. L. 2011. The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci, 32, 581-90.
PRICE, D. D. 2000. Psychological and neural mechanisms of the affective dimension of pain. Science, 288, 1769-72.
PRICE, D. D., VON DER GRUEN, A., MILLER, J., RAFII, A. & PRICE, C. 1985. A psychophysical analysis of morphine analgesia. Pain, 22, 261-9.
QUINTANA, P., SOTO, D., POIROT, O., ZONOUZI, M., KELLENBERGER, S., MULLER, D., CHRAST, R. & CULL-CANDY, S. G. 2015. Acid-sensing ion channel 1a drives AMPA receptor plasticity following ischaemia and acidosis in hippocampal CA1 neurons. J Physiol, 593, 4373-86.
RAINVILLE, P., DUNCAN, G. H., PRICE, D. D., CARRIER, B. & BUSHNELL, M. C. 1997. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science, 277, 968-71.
RAJA, S. N., CAMPBELL, J. N. & MEYER, R. A. 1984. Evidence for different mechanisms of primary and secondary hyperalgesia following heat injury to the glabrous skin. Brain, 107 ( Pt 4), 1179-88.
RAO, V. R. & FINKBEINER, S. 2007. NMDA and AMPA receptors: old channels, new tricks. Trends Neurosci, 30, 284-91.
RAYNOR, K., KONG, H., CHEN, Y., YASUDA, K., YU, L., BELL, G. I. & REISINE, T. 1994. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol, 45, 330-4.
REN, W., KIRITOSHI, T., GREGOIRE, S., JI, G., GUERRINI, R., CALO, G. & NEUGEBAUER, V. 2013. Neuropeptide S: a novel regulator of pain-related amygdala plasticity and behaviors. J Neurophysiol, 110, 1765-81.
REN, W. & NEUGEBAUER, V. 2010. Pain-related increase of excitatory transmission and decrease of inhibitory transmission in the central nucleus of the amygdala are mediated by mGluR1. Mol Pain, 6, 93.
REN, W. H., GUO, J. D., CAO, H., WANG, H., WANG, P. F., SHA, H., JI, R. R., ZHAO, Z. Q. & ZHANG, Y. Q. 2006. Is endogenous D-serine in the rostral anterior cingulate cortex necessary for pain-related negative affect? J Neurochem, 96, 1636-47.
REUVENY, E., SLESINGER, P. A., INGLESE, J., MORALES, J. M., INIGUEZ-LLUHI, J. A., LEFKOWITZ, R. J., BOURNE, H. R., JAN, Y. N. & JAN, L. Y. 1994. Activation of the cloned muscarinic potassium channel by G protein beta gamma subunits. Nature, 370, 143-6.
ROMANSKI, L. M., CLUGNET, M. C., BORDI, F. & LEDOUX, J. E. 1993. Somatosensory and auditory convergence in the lateral nucleus of the amygdala. Behav Neurosci, 107, 444-50.
RUSSELL, F. A., KING, R., SMILLIE, S. J., KODJI, X. & BRAIN, S. D. 2014. Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev, 94, 1099-142.
SAH, P., FABER, E. S., LOPEZ DE ARMENTIA, M. & POWER, J. 2003. The amygdaloid complex: anatomy and physiology. Physiol Rev, 83, 803-34.
SALVATORE, C. A., MALLEE, J. J., BELL, I. M., ZARTMAN, C. B., WILLIAMS, T. M., KOBLAN, K. S. & KANE, S. A. 2006. Identification and pharmacological characterization of domains involved in binding of CGRP receptor antagonists to the calcitonin-like receptor. Biochemistry, 45, 1881-7.
SANTORO, B., CHEN, S., LUTHI, A., PAVLIDIS, P., SHUMYATSKY, G. P., TIBBS, G. R. & SIEGELBAUM, S. A. 2000. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci, 20, 5264-75.
SARHAN, M., FREUND-MERCIER, M. J. & VEINANTE, P. 2005. Branching patterns of parabrachial neurons projecting to the central extended amgydala: single axonal reconstructions. J Comp Neurol, 491, 418-42.
SATO, M., ITO, M., NAGASE, M., SUGIMURA, Y. K., TAKAHASHI, Y., WATABE, A. M. & KATO, F. 2015. The lateral parabrachial nucleus is actively involved in the acquisition of fear memory in mice. Mol Brain, 8, 22.
SHARMA, S. K., NIRENBERG, M. & KLEE, W. A. 1975. Morphine receptors as regulators of adenylate cyclase activity. Proc Natl Acad Sci U S A, 72, 590-4.
SHIMADA, S., INAGAKI, S., NARITA, N. & TAKAGI, H. 1992. Synaptic contacts between CGRP-immunoreactive terminals and enkephalin-immunoreactive neurons in the central amygdaloid nucleus of the rat. Neurosci Lett, 134, 243-6.
SHIMADA, S., SHIOSAKA, S., EMSON, P. C., HILLYARD, C. J., GIRGIS, S., MACINTYRE, I. & TOHYAMA, M. 1985. Calcitonin gene-related peptidergic projection from the parabrachial area to the forebrain and diencephalon in the rat: an immunohistochemical analysis. Neuroscience, 16, 607-16.
SHOJI, Y., DELFS, J. & WILLIAMS, J. T. 1999. Presynaptic inhibition of GABA(B)-mediated synaptic potentials in the ventral tegmental area during morphine withdrawal. J Neurosci, 19, 2347-55.
SIMONIN, F., VALVERDE, O., SMADJA, C., SLOWE, S., KITCHEN, I., DIERICH, A., LE MEUR, M., ROQUES, B. P., MALDONADO, R. & KIEFFER, B. L. 1998. Disruption of the kappa-opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa-agonist U-50,488H and attenuates morphine withdrawal. Embo j, 17, 886-97.
SMITH, J. S., SCHINDLER, A. G., MARTINELLI, E., GUSTIN, R. M., BRUCHAS, M. R. & CHAVKIN, C. 2012. Stress-induced activation of the dynorphin/kappa-opioid receptor system in the amygdala potentiates nicotine conditioned place preference. J Neurosci, 32, 1488-95.
SORA, I., FUNADA, M. & UHL, G. R. 1997a. The mu-opioid receptor is necessary for [D-Pen2,D-Pen5]enkephalin-induced analgesia. Eur J Pharmacol, 324, R1-2.
SORA, I., TAKAHASHI, N., FUNADA, M., UJIKE, H., REVAY, R. S., DONOVAN, D. M., MINER, L. L. & UHL, G. R. 1997b. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci U S A, 94, 1544-9.
SUN, R. Q., LAWAND, N. B., LIN, Q. & WILLIS, W. D. 2004. Role of calcitonin gene-related peptide in the sensitization of dorsal horn neurons to mechanical stimulation after intradermal injection of capsaicin. J Neurophysiol, 92, 320-6.
SUN, R. Q., LAWAND, N. B. & WILLIS, W. D. 2003. The role of calcitonin gene-related peptide (CGRP) in the generation and maintenance of mechanical allodynia and hyperalgesia in rats after intradermal injection of capsaicin. Pain, 104, 201-8.
SUWANPRATHES, P., NGU, M., ING, A., HUNT, G. & SEOW, F. 2003. c-Fos immunoreactivity in the brain after esophageal acid stimulation. Am J Med, 115 Suppl 3A, 31s-38s.
SVOBODA, K. R. & LUPICA, C. R. 1998. Opioid inhibition of hippocampal interneurons via modulation of potassium and hyperpolarization-activated cation (Ih) currents. J Neurosci, 18, 7084-98.
TAKAHASHI, T., SVOBODA, K. & MALINOW, R. 2003. Experience strengthening transmission by driving AMPA receptors into synapses. Science, 299, 1585-8.
TANIMOTO, S., NAKAGAWA, T., YAMAUCHI, Y., MINAMI, M. & SATOH, M. 2003. Differential contributions of the basolateral and central nuclei of the amygdala in the negative affective component of chemical somatic and visceral pains in rats. Eur J Neurosci, 18, 2343-50.
THOMAS, D. A., OLIVERAS, J. L., MAIXNER, W. & DUBNER, R. 1992. Systemic morphine administration attenuates the perceived intensity of noxious heat in the monkey. Pain, 49, 129-35.
TIAN, M., BROXMEYER, H. E., FAN, Y., LAI, Z., ZHANG, S., ARONICA, S., COOPER, S., BIGSBY, R. M., STEINMETZ, R., ENGLE, S. J., MESTEK, A., POLLOCK, J. D., LEHMAN, M. N., JANSEN, H. T., YING, M., STAMBROOK, P. J., TISCHFIELD, J. A. & YU, L. 1997. Altered hematopoiesis, behavior, and sexual function in mu opioid receptor-deficient mice. J Exp Med, 185, 1517-22.
TURNER, B. H. & HERKENHAM, M. 1991. Thalamoamygdaloid projections in the rat: a test of the amygdala’s role in sensory processing. J Comp Neurol, 313, 295-325.
TWITCHELL, W. A. & RANE, S. G. 1993. Opioid peptide modulation of Ca(2+)-dependent K+ and voltage-activated Ca2+ currents in bovine adrenal chromaffin cells. Neuron, 10, 701-9.
UEDA, T., UGAWA, S., SAISHIN, Y. & SHIMADA, S. 2001. Expression of receptor-activity modifying protein (RAMP) mRNAs in the mouse brain. Brain Res Mol Brain Res, 93, 36-45.
UNTERWALD, E. M., KNAPP, C. & ZUKIN, R. S. 1991. Neuroanatomical localization of kappa 1 and kappa 2 opioid receptors in rat and guinea pig brain. Brain Res, 562, 57-65.
VAN’T VEER, A. & CARLEZON, W. A., JR. 2013. Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology (Berl), 229, 435-52.
VAN ROSSUM, D., HANISCH, U. K. & QUIRION, R. 1997. Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci Biobehav Rev, 21, 649-78.
VANDERAH, T. W. 2010. Delta and kappa opioid receptors as suitable drug targets for pain. Clin J Pain, 26 Suppl 10, S10-5.
VANDERAH, T. W., LARGENT-MILNES, T., LAI, J., PORRECA, F., HOUGHTEN, R. A., MENZAGHI, F., WISNIEWSKI, K., STALEWSKI, J., SUEIRAS-DIAZ, J., GALYEAN, R., SCHTEINGART, C., JUNIEN, J. L., TROJNAR, J. & RIVIERE, P. J. 2008. Novel D-amino acid tetrapeptides produce potent antinociception by selectively acting at peripheral kappa-opioid receptors. Eur J Pharmacol, 583, 62-72.
VANDERAH, T. W., SCHTEINGART, C. D., TROJNAR, J., JUNIEN, J. L., LAI, J. & RIVIERE, P. J. 2004. FE200041 (D-Phe-D-Phe-D-Nle-D-Arg-NH2): A peripheral efficacious kappa opioid agonist with unprecedented selectivity. J Pharmacol Exp Ther, 310, 326-33.
VAUGHAN, C. W., INGRAM, S. L., CONNOR, M. A. & CHRISTIE, M. J. 1997. How opioids inhibit GABA-mediated neurotransmission. Nature, 390, 611-4.
VEINANTE, P., YALCIN, I. & BARROT, M. 2013. The amygdala between sensation and affect: a role in pain. J Mol Psychiatry, 1, 9.
VERTES, R. P. & HOOVER, W. B. 2008. Projections of the paraventricular and paratenial nuclei of the dorsal midline thalamus in the rat. J Comp Neurol, 508, 212-37.
VLAEYEN, J. W. 2015. Learning to predict and control harmful events: chronic pain and conditioning. Pain, 156 Suppl 1, S86-93.
VOGT, B. A., ROSENE, D. L. & PANDYA, D. N. 1979. Thalamic and cortical afferents differentiate anterior from posterior cingulate cortex in the monkey. Science, 204, 205-7.
WALKER, C. S., CONNER, A. C., POYNER, D. R. & HAY, D. L. 2010. Regulation of signal transduction by calcitonin gene-related peptide receptors. Trends Pharmacol Sci, 31, 476-83.
WANG, M., RAMOS, B. P., PASPALAS, C. D., SHU, Y., SIMEN, A., DUQUE, A., VIJAYRAGHAVAN, S., BRENNAN, A., DUDLEY, A., NOU, E., MAZER, J. A., MCCORMICK, D. A. & ARNSTEN, A. F. 2007. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell, 129, 397-410.
WATABE, A. M., OCHIAI, T., NAGASE, M., TAKAHASHI, Y., SATO, M. & KATO, F. 2013. Synaptic potentiation in the nociceptive amygdala following fear learning in mice. Mol Brain, 6, 11.
WATKINS, L. R. & MAYER, D. J. 1982. Organization of endogenous opiate and nonopiate pain control systems. Science, 216, 1185-92.
WAYMAN, G. A., LEE, Y. S., TOKUMITSU, H., SILVA, A. J. & SODERLING, T. R. 2008. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron, 59, 914-31.
WHITEHEAD, G., JO, J., HOGG, E. L., PIERS, T., KIM, D. H., SEATON, G., SEOK, H., BRU-MERCIER, G., SON, G. H., REGAN, P., HILDEBRANDT, L., WAITE, E., KIM, B. C., KERRIGAN, T. L., KIM, K., WHITCOMB, D. J., COLLINGRIDGE, G. L., LIGHTMAN, S. L. & CHO, K. 2013. Acute stress causes rapid synaptic insertion of Ca2+ -permeable AMPA receptors to facilitate long-term potentiation in the hippocampus. Brain, 136, 3753-65.
WICKMAN, K. D., INIGUEZ-LLUHL, J. A., DAVENPORT, P. A., TAUSSIG, R., KRAPIVINSKY, G. B., LINDER, M. E., GILMAN, A. G. & CLAPHAM, D. E. 1994. Recombinant G-protein beta gamma-subunits activate the muscarinic-gated atrial potassium channel. Nature, 368, 255-7.
WILEY, J. W., GROSS, R. A. & MACDONALD, R. L. 1992. The peptide CGRP increases a high-threshold Ca2+ current in rat nodose neurones via a pertussis toxin-sensitive pathway. J Physiol, 455, 367-81.
WILLIAMS, J. T., CHRISTIE, M. J. & MANZONI, O. 2001. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev, 81, 299-343.
WILLIAMS, J. T., INGRAM, S. L., HENDERSON, G., CHAVKIN, C., VON ZASTROW, M., SCHULZ, S., KOCH, T., EVANS, C. J. & CHRISTIE, M. J. 2013. Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev, 65, 223-54.
WINTERS, B. L., GREGORIOU, G. C., KISSIWAA, S. A., WELLS, O. A., MEDAGODA, D. I., HERMES, S. M., BURFORD, N. T., ALT, A., AICHER, S. A. & BAGLEY, E. E. 2017. Endogenous opioids regulate moment-to-moment neuronal communication and excitability. Nat Commun, 8, 14611.
WOOLF, C. J. 1983. Evidence for a central component of post-injury pain hypersensitivity. Nature, 306, 686-8.
WOOLF, C. J. 2011. Central sensitization: implications for the diagnosis and treatment of pain. Pain, 152, S2-15.
WU, Z. Z., CHEN, S. R. & PAN, H. L. 2004. Differential sensitivity of N- and P/Q-type Ca2+ channel currents to a mu opioid in isolectin B4-positive and -negative dorsal root ganglion neurons. J Pharmacol Exp Ther, 311, 939-47.
XU, W., LUNDEBERG, T., WANG, Y. T., LI, Y. & YU, L. C. 2003. Antinociceptive effect of calcitonin gene-related peptide in the central nucleus of amygdala: activating opioid receptors through amygdala-periaqueductal gray pathway. Neuroscience, 118, 1015-22.
YIZHAR, O., FENNO, L. E., DAVIDSON, T. J., MOGRI, M. & DEISSEROTH, K. 2011. Optogenetics in neural systems. Neuron, 71, 9-34.
YOUNG, A. W., AGGLETON, J. P., HELLAWELL, D. J., JOHNSON, M., BROKS, P. & HANLEY, J. R. 1995. Face processing impairments after amygdalotomy. Brain, 118 ( Pt 1), 15-24.
ZHANG, F., WANG, L. P., BRAUNER, M., LIEWALD, J. F., KAY, K., WATZKE, N., WOOD, P. G., BAMBERG, E., NAGEL, G., GOTTSCHALK, A. & DEISSEROTH, K. 2007. Multimodal fast optical interrogation of neural circuitry. Nature, 446, 633-9.
ZHANG, R. X., ZHANG, M., LI, A., PAN, L., BERMAN, B. M., REN, K. & LAO, L. 2013. DAMGO in the central amygdala alleviates the affective dimension of pain in a rat model of inflammatory hyperalgesia. Neuroscience, 252, 359-66.
ZHANG, X. J., ZHANG, T. W., HU, S. J. & XU, H. 2011a. Behavioral assessments of the aversive quality of pain in animals. Neurosci Bull, 27, 61-7.
ZHANG, Y., WANG, N., WANG, J. Y., CHANG, J. Y., WOODWARD, D. J. & LUO, F. 2011b. Ensemble encoding of nociceptive stimulus intensity in the rat medial and lateral pain systems. Mol Pain, 7, 64.
ZHANG, Y. P. & OERTNER, T. G. 2007. Optical induction of synaptic plasticity using a light-sensitive channel. Nat Methods, 4, 139-41.
ZHAO, S., CUNHA, C., ZHANG, F., LIU, Q., GLOSS, B., DEISSEROTH, K., AUGUSTINE, G. J. & FENG, G. 2008. Improved expression of halorhodopsin for light-induced silencing of neuronal activity. Brain Cell Biol, 36, 141-54.
ZHU, W. & PAN, Z. Z. 2004. Synaptic properties and postsynaptic opioid effects in rat central amygdala neurons. Neuroscience, 127, 871-9.
ZHU, W. & PAN, Z. Z. 2005. Mu-opioid-mediated inhibition of glutamate synaptic transmission in rat central amygdala neurons. Neuroscience, 133, 97-103.
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Biomedical Science"
Biomedical Science focuses on how cells, organs and systems function in the human body and underpins much of modern medicine. Biomedical Science applies parts of natural and/or formal sciences to help develop advances in healthcare.
Related Articles

DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: