Targeted Drug Delivery: Enhanced Permeability and Retention Effect and Nano Technology
Info: 8827 words (35 pages) Dissertation
Published: 9th Dec 2019
Tagged: MedicalMedical Technology
Obstacles standing in cancer cure or efficient treatment include: 1) lack of early diagnosis; 2) cancer stem cells and metastasis; 3) great cytotoxicity to cancer cells while low toxicity to normal cells; 4) prolonged tumor accumulation. A tumor targeting drug delivery system could solve or overcome these obstacles from 1) to 4). In general, there are two types of targeting strategies: passive targeting and active targeting. Passive targeting is based on biophysiological properties (leaky of tumor blood vessel and lack of lymph drain) of tumor. It utilizes an effect called enhanced permeability and retention effect to deliver large molecules (size of 10-100 nm) to tumor tissue. On the other hand, active targeting takes advantage of targeting moieties like antibody or ligands to deliver drug into cells. When the monoclonal antibodies (mAbs) or ligands bind with antigens or ligand receptors overexpressed on the surface of cancer cells, the whole molecules/conjugates will internalize into the cells.
4.1.1 EPR effect and Nano technology in drug delivery
International Union of Pure and Applied Chemistry (IUPAC) defined nanoparticles as particle of any shape with size in the 1–100 nm range.1 Nanotechnology enabled effective and safe treatment for numerous diseases, especially cancer, and lead to a new category of drug, nanomedicine. Some distinguishing characteristic of nanomedicines including: 1) site specific delivery of drugs at pathological sites (tissue); 2) enhanced pharmaceutical properties, such as increased stability, solubility; 3) extended circulation times by decreasing excretion or degradation by renal or liver; 4) improved therapeutic index by increasing accumulation in target tissue and reducing toxicities against healthy organ; 5) co-delivery of two or more drugs as combination therapy.1,2 Besides, nanomedicines enable stimulus-triggered drug release and delivery of macromolecular drugs like DNA, RNA and protein. More interestingly, nanomedicines for cancer treatment facilitate visualization of tumor by combining imaging molecules with drugs, referred as theranostics.3 However, lack of optimized targeting parameters stands as a key challenge for nanomedicines’ development.4 Recently, functionalized nanoparticles to deliver therapeutic drug to certain cancer cells expressing specific molecular target (active targeting nanoparticles) have been utilized as a strategy to enhance targeting specificity.4
Generally therapeutic nanoparticles are injected intravenously, and they are likely to accumulate in tumor by the enhanced permeability and retention (EPR) effect.5 Given the rapid growth of solid tumor and their high demand of nutrition and oxygen, cancer cells stimulate the formation of blood vessels by releasing vascular endothelial growth factor (VEGF) and some other growth factors. The resulted tumor blood vessel cells exhibit defective morphology and aberrant architecture with wide fenestrations, different from the normal healthy vascular.6 The poorly aligned leaky tumor blood vessel enabled extravasation of nanoparticles. Additionally, lack of efficient lymphatic drainage in tumor tissue confines the removal of the nanoparticles. These two pathways work together to contribute to the enhanced permeability and retention (EPR) effect.6 EPR effect induced drug targeting is based on the pathological properties of tumor tissue, thus, it is considered as passive drug targeting.
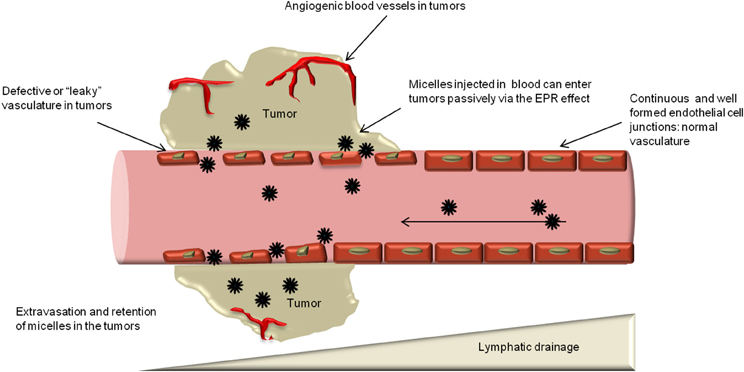
Figure 4‑1. EPR effect
A variety of nanoparticles have been created and evaluated these years. The nanocarrier platforms include liposomes, polymers, micells, dendrimers, protein based nanoparticle (ADC), polymer-drug conjugate and inorganic nanoparticles.
Liposomes
Doxil, a PEGylated liposomal formulation of doxorubicin, was the first therapeutic nanoparticle being approved by FDA for cancer treatment.7 Liposomes are phospholipids bilayer forming spherical closed-colloidal vesicles in aqueous solution.8 Liposome’s ability to encapsulate hydrophobic drugs inside of the lipid bilayers and hydrophilic drugs in the interior aqueous compartment has made it an attractive candidate as nanocarrier.9 Liposomes exhibited good pharmacokinetics and distribution in the body due to their high drug loading percentage, high stability and biocompatibility.10 Further introduction of hydrophilic polyethylene glycol (PEG) onto exterior of liposomes (stealth liposomes) help them to evade immune system, especially the mononuclear phagocyte system. As a result, the circulation time of PEG coated liposomes has been prolonged.11 Improved distribution and accumulation of liposome drugs leads to increased activity and decreased toxicity.12 The drug release in liposome could be triggered by either environmental factors like pH, enzyme, or external factors like heat or ultrasonic.13 The diversity of liposome structure and composition allows introducing imaging molecules into liposome, which could be applied in magnetic resonance imaging (MRI)14 and computed tomography (CT)15. Till now, liposomes have been the most successful nanoparticle delivery system for a lot diseases including cancer.
Biodegradable polymeric nanoparticles
Poly-D,L-lactide-co-glycolic acid (PLGA), polylactic acid (PLA), poly-ɛ-caprolactone (PCL), poly-alkyl- cyanoacrylates (PACA), chitosan and gelatin are the most commonly used polymeric nanoparticles with outstanding biocompatibility, resorbability, and muco-adhesive characteristics.16 The therapeutic agents including large DNA are usually linked, diffused, adsorbed, entangled, or encapsulated into the polymeric matrix.17 Biodegradable polymeric nanoparticles are favored for their subcellular size, controlled drug release, stability and nontoxicity.18 These biodegradable polymeric nanoparticles are compatible with cells and tissues, they are not likely to generate thrombosis, or lead to inflammatory. Also, polymeric nanoparticles are able to avoid reticuloendothelial system and not induce immune reaction. 19 The drug release mechanisms of polymeric nanoparticles could be diffusion, degradation, burst or a combination of above. 16
Polymeric Micelles
Surfactant molecules with hydrophilic head and hydrophobic tail undergo self-assembly in aqueous solution to aggregate and generate compositions with a stable hydrophilic shell.20 Thus, micelles provide a great opportunity for delivery of hydrophobic therapeutic agents for cancer treatment, especially poorly water soluble ones.20 The relatively smaller size of micelles benefits micelles’ transportation by either percutaneous lymphatic system or leaky blood vessels through EPR effect.21 Meanwhile, the size of micelles is large enough to disseminate variety of materials such as small molecule drug, protein, DNA, etc. When compared with liposomes, micelles are sterically more stable and show prolonged circulation time.21 The shape of micelles varies from spheres, rods, vesicles, tubules, and lamellae depending on the ratio of hydrophobic/hydrophilic blocks and solvent environment. 22,23 The shape of micelles has an essential impact on the pharmacokinetic properties, especially circulation time.24 The frequently used polymeric systems involve poly(lactic acid) (PLA), poly (l-lysine) (PLL) and poly (beta-amino ester) as hydrophobic domain and poly (ethylene glycol) (PEG), poly(N-vinyl pyrrolidone) (PVP) and poly(N-isopropyl acrylamide) (pNIPAM) as hydrophilic domain.25 Micelle nanoparticles increase the solubility of drugs, enhance drugs’ accumulation in target tissue and decrease the toxicity in in vivo test. Genexol-PM is a PEG-PDLLA copolymer micelle formulation of taxol,26 it has been approved by FDA as anticancer nanomedicine. In the preclinical study, Genexol-PM lead to 3 times higher concentration in tumor tissue than paclitaxel itself.27 Mixing imaging aspects with therapeutic drugs make visualization of micelle pathway possible.3
Dendrimers
Dendrimers’ well defined structure and easy-to-functionalize features show great potential as a drug delivery system. Dendrimers allows entrapping or conjugating therapeutic drugs by either host-guest interaction or covalent bond.28 Dendrimers are symmetric branched polymers with spherical 3-D conformation. Dendrimers consist of a central core, an inner shell and an outer shell. 29 The most commonly used core includes polyamidoamine (PAMAM), ethylene diamine (EDA), polypropylimine (PPI) and ethylene diamine (EDA).30 The exterior functional group can be modified to charged species, carboxyl, amine, or alcoholic groups to benefit dendrimers’ solubility.31 The layers between each cascade point are defined “Generations.” With generation increasing, dendrimers’ size and number of functional group amplifies to provide more reactions site, which is termed as highly functional.29 Nowadays, the dendrimer–drug conjugation is achieved by a covalent bond between an anticancer drug and a surface group of dendrimer. In this way, therapeutic drug like paclitaxel, doxorubicin, methotrexate, etc., and targeting entities such as folic acid, biotin, could be loaded onto dendrimer through ester or amide bond.28 Interestingly, the cationic nature and high ratio of surface group to volume enables PAMA dendrimer’s binding to DNA; make dendrimer a suitable delivery vehicle for DNA.32 However, the cationic dendrimer macromolecules are likely to interact with negative charged cell membranes and lead to cell lysis.33 Functionalize the surface groups in dendrimer or change them to anionic results in drastic reduction of toxicity. 34
Inorganic Nanoparticles
Comparing with previous mentioned liposome, polymeric nanoparticles and micelles, inorganic nanoparticles are generally smaller in size (5-100 nm) and inflexible. Basically, inorganic nanoparticle is composed of an inorganic core (quantum dots, carbon nanotubes, gold, iron oxide, silica, etc.) and a biodegradable shell to increase solubility and inhibit aggregation. Quantom dots consist of 10-50 cadmium or mercury atoms with 2-10 nm diameter.35 Their ability to discrete narrow frequencies, stability and brightness allow their application in tumor imaging.36 Carbon nanotubes are comprised of hexagonal arrangement of carbon atoms with fixed C-C distance of 1.4 Ǻ and form an open-ended or capped cylinder conformation.37 The carbon nanotubes are able to be functionalized by either covalent38 or noncovalent bond39. Their needle-like shape benefits their penetration into cells without obvious damage though the mechanism is unclear.37 Gold nanoparticles are easy to be functionalized by amine and thiol groups, which distinguish them as great candidates for medical applications.40 Besides application in drug delivery, gold nanoparticles can be used in radiation therapy because of their photo-thermal and radioactive properties.41 Iron oxide nanoparticles refer to magnetite (Fe3O4) or maghemite (Fe2O3) core with surface layer of albumin, PEG, or dextran.42 Their magnetic properties aids prolonged retention time in tumor with extra magnetic field, 43 and they are visible under MRI44. Ceramic nanoparticles such as silica show some biocompatibility but the biodegradability is still a concern. The silica nanoparticles delivering gene to spleen are able to trigger immune response to attack target tumor tissue.45
Others
Polymer-drug conjugates enhance drugs’ pharmacokinetics by slowing down drugs’ digestion and extending the circulation time.46 Meanwhile, conjugating water soluble polymer with drugs improves drugs’ solubility.47 Besides polymer, antibody is also employed as a nanoparticle to conjugate with drug. Albumin conjugated drug was also found to improve solubility, decrease toxicity and increase drugs’ concentration in tumor site by targeting albumin receptor (gp60).48
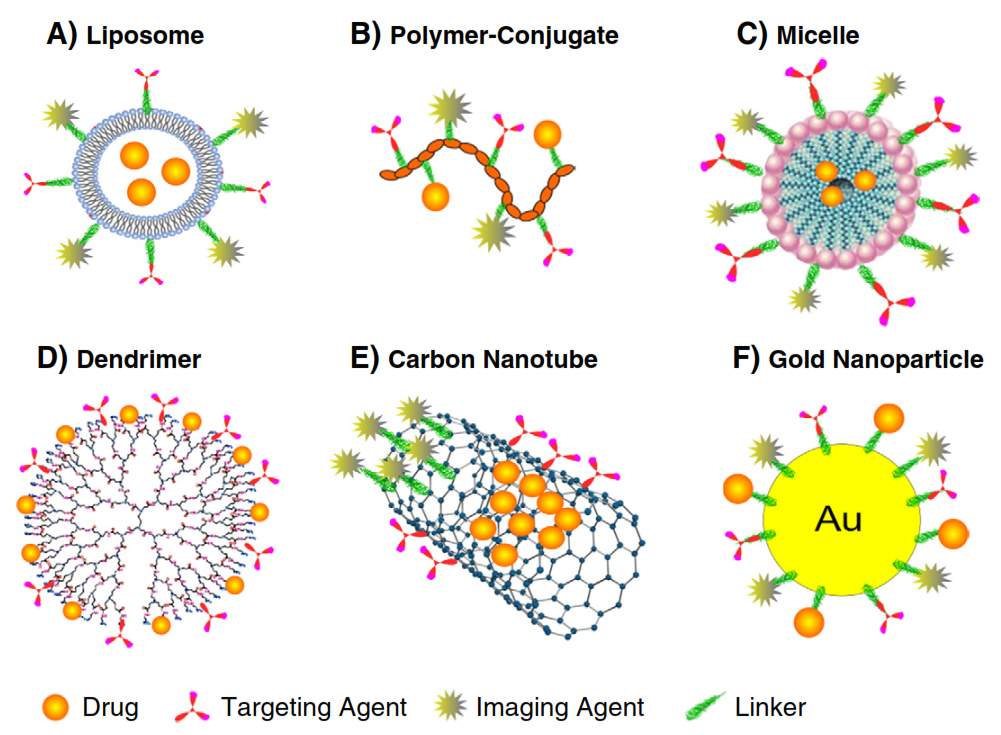
Figure 4‑2. Cartoon illustration of nanoparticles drug delivery system
Several therapeutic nanomedicines have been approved and commercialized utilizing platforms such as liposomes, albumin nanoparticles, polymer conjugates and micelles. The active pharmaceutical ingredients applied nanotechnologies for cancer treatment include doxorubicin, daunorubicin, vincristine, irinotecan, paclitaxel, etc. Doxil, a PEGylated liposome formulation of doxorubicin, exhibits a long circulation halflife of approximately 2 days.49 DaunoXome increased tumor uptake of daunorubicin to 10 fold comparing with free drug, and reduced cardiotoxicity of daunorubicin.50 Marqibo is liposomal vincristine with increased circulation time and tumor retention. When used as a component in combination treatment against pancreatic cancer, Onivyde (a liposome formulation of irinotecan) improved overall survival (OS) from 4.2 months to 6.2 months.51 Myocet as a liposomal formulation of doxorubicin exhibit similar anticancer activity with free drug, but reduce drug related cardiotoxicity.52 Mepact is a liposomal formulation of mifamurtide, prevent free drugs’ rapid clearance from the body.53 Abraxane is albumin-bound paclitaxel; this formulation increases therapeutic index, decreases infusion time, and improves pharmacokinetic parameters of paclitaxel.54 SMANCS is a conjugate of polymeric styrene maleic acid (SMA) and anticancer drug neocarzinostatin (NCS), severe adverse reactions have been reported in treatment of hepatocellular carcinoma in 2002.55 Genexol-PM ,a micelle formulation of paclitaxel, release free drug in a controlled manner.56 In the Genexol-PM treatment against advanced non-small cell lung cancer, overall survival rate of 14.8 months has been observed and adverse effect caused by Cremorphor EL has been avoided.57
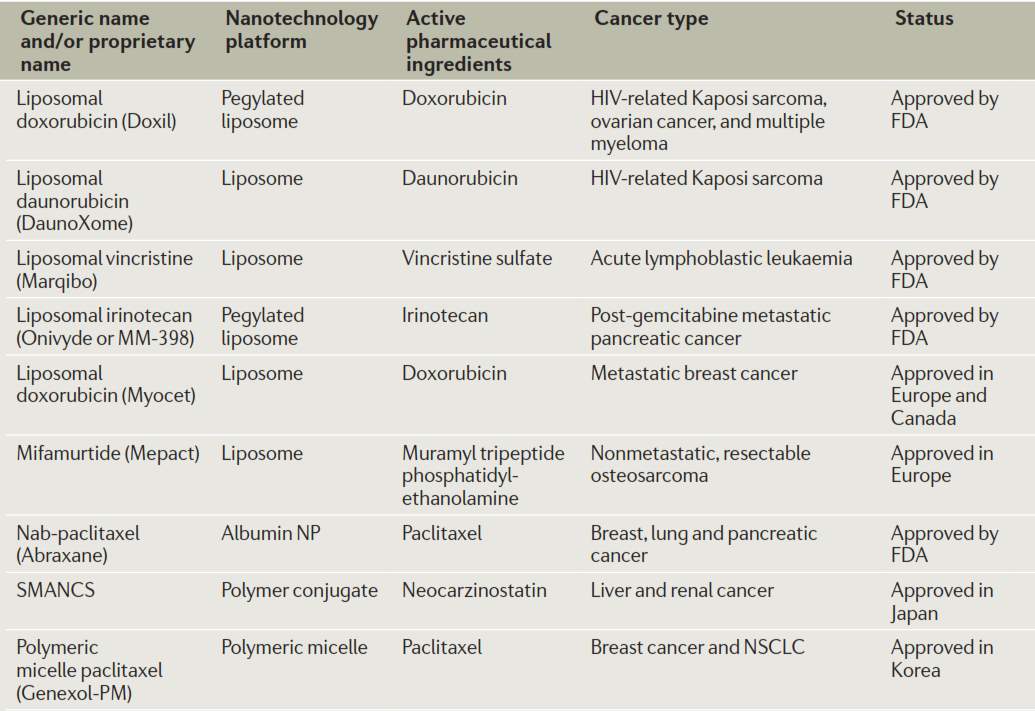
Figure 4‑3. Commercialized nanomedicines
Besides above mentioned FDA approved Abraxane and Korea FDA approved Genexol-PM, several other paclitaxel nanomedicine have been evaluated in clinical trials including Opaxio58 (paclitaxel poliglumex, phase 3) for ovarian cancer treatment; endoTAG-159 (cationic liposomal paclitaxel, phase 3) for metastatic triple-negative breast cancer treatment; LEP-ETU60 (liposomal encapsulated paclitaxel, phase 2) for ovarian cancer treatment; and Paclical61 (paclitaxel-loaded micelles, phase 3) for ovarian cancer treatment.
4.1.2 Gold nanoparticles in cancer treatment
Gold nanoparticle as drug delivery system
Lots of inorganic nanoparticles, especially gold nanoparticles, have become promising platforms for drug delivery. Gold nanoparticles are inert and stable in biological system, and they did not show significant toxicity in the body.62 Given the ease of Au-S bond formation, gold nanoparticles can be functionalized with surfacing ligand through stable gold-thiol bond.63 Also, gold nanoparticles with different sizes ranging from 1 nm to 150 nm are accessible.64 All above mentioned properties of gold nanoparticle allow its application in drug delivery. Additionally, gold nanoparticles could be loaded with therapeutic drug for controlled release.65 The stimuli could be internal, such as pHof tumor microenvironment, 66 or enzyme overexpressed in the cancer cells;67 or external factors, like light68, or heat, etc. With long time accumulation of gold nanoparticles in tumor tissue, the drug could be released gradually and maintain the concentration within therapeutic window for extended time.66
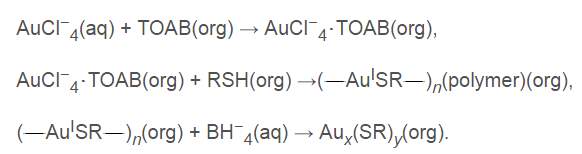
Figure 4‑4. Scheme of gold nanoparticles synthesis
As shown in Fig. 4-4, gold nanoparticles are formed and stabilized in the organic solution in presence of alkanethiol ligands. Sodium citrate serves as a reducing agent and a stabilizer to prevent aggregation in synthesis.69 This technique facilitates rapid synthesis of monodispersed gold nanoparticles capped by compressed surfacing group. The surface layer displays an important role of functionality depending on the terminal group, such as amine, carboxyl, to alcoholic group, etc. Other than targeting agents, polyethylene glycol (PEG) could be introduced onto the surface of gold nanoparticles to increase the stability, solubility and circulation time.70 Currently, a tumor necrosis factor α TNF-bound gold nanoparticle (CYT-6091) is under investigation in phase 1 clinical trial against advanced solid tumors. CYT-6091 is well tolerated in the body, even at highest treated dose (3 times of maximum tolerated dose of TNF), no dose-limiting toxic effect was observed. 71 The gold nanoparticles prolonged circulation time of TNF from 28 min to 130 min.71
Physicochemical characteristic of gold nanoparticles such as coating ligand and size would affect their toxicity, cellular uptake, distribution significantly. For example, PEGylated gold nanoparticles showed longer blood circulation time than non-coated ones, while further introducing galactose-PEG-thiols (targeting hepatocytes) onto gold nanoparticles improved Gal-PEG-NPs’s retention in liver. Comparing different size of gold nanoparticles, 50 nm Gal-PEG grafted ones exhibited best site-specificity. 72 Generally, gold nanoparticles were nontoxic at up to 250 nM concentration in human cells.73 The toxicity and biodistribution problems are complicated that no relationship between size and toxicity can be concluded now.74 Zhang, et.al found that the PEG-coated gold nanoparticles did not lead to obvious body weight decrease in vivo. They also reported PEGylated gold nanoparticles with 10 nm, 60 nm diameters are more toxic than 5 nm and 30 nm ones since 10 nm and 60 nm AuNPs upregulated alanine transaminase and creatinine level dramatically, indicating liver and kidney toxicity.74 Biodistribution of gold nanoparticles varies with size too. Overall, small gold nanoparticles with size of 5-15 nm exhibited a wilder organ distribution than larger 50-100 nm ones, and liver and spleen are two dominant organs for AuNPs’ distribution and metabolism.75 Gold nanoparticles with PEG coating decrease RES uptake, and reduce the charge-based proteins-gold interaction, thus they are less likely to accumulate in liver (one third amount of the uncoated ones).76 Liver is the dominant organ for smaller PEGylated gold nanoparticles (5-10 nm) while spleen is the dominant organ for larger ones (30 nm). Size up to 60 nm PEGylated AuNPs showed a low distribution in organs, indicating their metabolism by liver and kidney.74 Zhang et.al concluded that 30 nm PEGylated gold nanoparticles are safe.74
Gold nanoparticles as radio sensitizer
Gold shows great absorption of photons and it can release secondary energy for such as X-ray, photoelectrons, and auger electrons to the surrounding tissue under radiation exposure.77 Thus, AuNPs are suitable candidates for radiosensitizer. Gold nanoparticles serve as radiosensitizer by producing reactive oxygen species (ROS), inducing oxidative stress, repairing damaged DNA, disrupting cell cycle, and some bystander effect.78 Despite of inertness of gold atom, the electronically active surface enables electron transfer from gold nanoparticles to oxygen and generate superoxide, lead to ROS production.79 Furthermore, the generated ROS will oxidize mitochondrial membrane80 and result in superoxide anions’ leaking into cells, and further damage the DNA.81 Also, AuNPs alters cell kinetics82 and shift cell cycle distribution to G2/M phase which is most radiosensitive.83 Radiation therapy induces DNA’s double strand breaks, and DNA’s repair is very important for cell survival. In presence of gold nanoparticles, the repair procedure is delayed or inhibited, which contribute to AuNPs’ radiosensitization.84 Bystander effect described the phenomenon that cells not directly exposed to radiation still behave in the similar manner as direct exposed ones.85 Gold nanoparticles can mediate bystander effect to the bulky cells include endothelial, fibroblast, lymphocytes in tumor microenvironment after radiation therapy.86 For example, after gold nanoparticles and radiation treatment of small airway epithelial cells (SAECs), the nearby lung fibroblasts upregulated 47 proteins and downregulated 62 proteins related to cell adhesion and extracellular matrix in response to the bystander effect.87
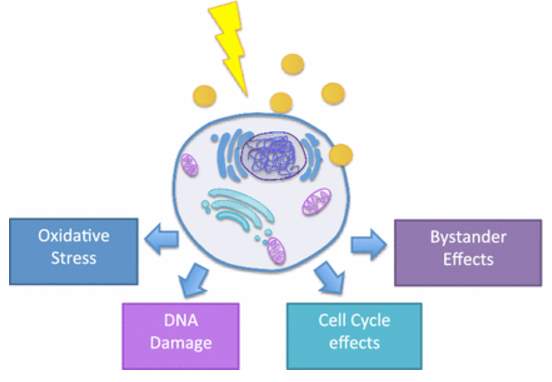
Figure 4‑5. Targeted gold nanoparticles as radiosensitizer
As early as Wilhelm Roentgen generate first human X-ray image, gold’s ability to induce strong X-ray attenuation was demonstrated. Different tissues provide distinguish level of X-ray attenuation to form X-ray based CT (computed tomography) image. The attenuation coefficient is a factor to describe how easy the material is to be penetrated by X-ray, and it is determined by atomic number and electron density of the tissue. Larger attenuation coefficient indicates attenuation of X-ray. Gold, with atomic number of 79 and electron density of 19.32 g/cm3, gives better signal than human tissue and even current contrast agent Iodine (53 and 4.9 g/cm3). For CT scan signal, the shape and size of gold nanoparticle is not crucial, but the number of AuNPs per volume matters. Previously, Hainfeld et. al utilized gold nanoparticles to induce vascular contrast enhancement in CT imaging in the in vivo test.88 Lately, PEG coated gold nanoparticles89 and immuno-targeted gold nanoparticles90 have been developed as CT contrast agent. These targeted gold nanoparticles allow cancer detection at cellular level by CT. 30 nm gold nanoparticles were coated by PEG5000 (non-targeted AuNPs)91, or further conjugated with anti-EGFR mAb Erbitux by EDC coupling reaction (targeted AuNPs).92 No obvious differences of AuNPs accumulation were observed at first 3 hours. In the time period of 3-6 hours after injection, targeted AuNPs were accumulating steadily in tumor tissue while non-targeted ones were cleared from the tumor gradually. The CT numbers of targeted AuNPs reached maximal accumulation after 6 hours post injection.92
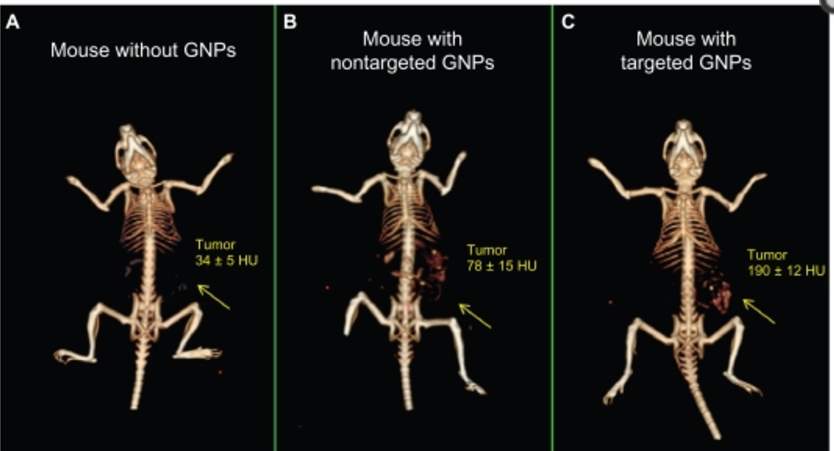
Figure 4‑6. CT image of mice injected with gold nanoparticles
Passive targeting mechanism of non-targeted gold nanoparticles enables their accumulation in leaky tumor tissue, but they are rapidly cleared from tumor and accumulate in kidney, liver and spleen93 at the time point of 6 hours after gold nanoparticles’ injection. Meanwhile, active targeting gold nanoparticles improved their uptake by cancer cell and prolonged their retention time. The large amount of targeted gold nanoparticles and their extended accumulation in cancer cells overcame the low sensitivity of CT scan and provide a great strategy to track tumor in body.92
4.1.3 Targeted drug delivery
Comparing active targeting and passive targeting mentioned above, active targeting of anticancer therapeutics shows advantages in affinities and internalization in tumors.94 The targeting ligands including antibody, sugar, and protein is recognized by cancer cells’ surface receptor, and thus extend drugs’ accumulation and residence time in tumor.95 Targeted drug delivery could be achieved in two approaches: attachment of macromolecule antibody/mAb, or small tumor-targeting molecules (TTM). Kadcyla® is a famous example of antibody-drug conjugate, targeting breast cancer cells overexpresse HER2 receptor and deliver theraputic agent emtansine (DM1).96 Taxoprexin, a DHA conjugated paclitaxel is also under Phase 1/2 investigation.97
The internalization of targeted drug delivery system is based on receptor mediated endocytosis. After the ligand bind to the cell surface receptor, it diffuse toward the cell membrane until it meet clathrin-coated pit,98 and then the pit will be pinch off from membrane as a vehicle to be captured by endosome. Although there are some other mechanisms, clathrin-mediated endocytosis is the most well studied one. The receptor mediated drug delivery is impacted by following factors: 1) how fast the ligand bind to receptor; 2) how fast the receptor recycle back to the surface; 3) how fast drug release; 4) receptor amount and saturation level.99 The receptor expression level and internalization parameters vary in different cells.
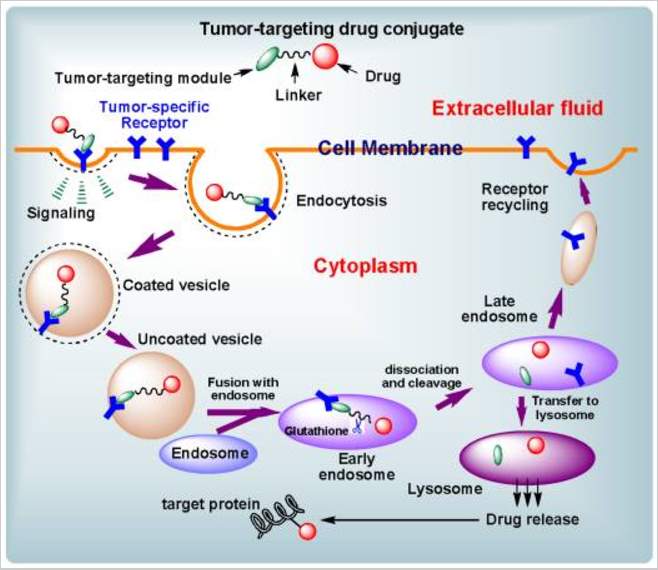
Figure 4‑7. Cartoon of receptor mediated endocytosis
However, these antibody-drug conjugates (ADCs) or ligand-drug conjugates have their deficiency. Most mAbs are too large to leak through tumor blood vessel and diffuse into tumors.100 And for the ADCs entered tumor tissue, they are trapped by perivascular tumor cells, but not penetrate into tumor mass, described as antigen barrier.101 Meanwhile, for the ADCs not enter tumor tissue, their slow clearance lead to prolonged accumulation in healthy tissue.102 Additionally, mAb’s immunogenic properties need to be taken into consideration when it is used as drug delivery system.103 The commonly used small-molecule targeting moieties including vitamins, fatty acid, hormones, etc. show high binding affinity with their receptors.104 Combining small molecule targeting and nanotechnology together lead to a drug delivery system with improved efficiency through highly specific tumor targeting and increased intracellular uptake.105
4.1.4 Biotin and biotin receptor
Living cells especially cancer cells need vitamins for their survival.106 The fast growing cancer cells require much more vitamins than normal ones due to its enhanced proliferation.106 Thus, the vitamin receptors related to vitamin uptake were found to be overexpressed on the surface of certain cancer cells. These vitamin receptors include B12, folate, and biotin receptors show great potential as therapeutic targets for anticancer drug delivery and as biomarkers for cancer cell imaging and identification.106 Among them, folate receptor has been studied extensively and lead to generation of some FDA approved targeted chemotherapy agent.7 Biotin has been utilized as targeting molecule for cancer treatment after Russell-Jones reported its high expression in variety of cancer cells in 2004.106 Biotin is a water soluble vitamin involves in regulation of epigenetics,107 synthesis and/or metabolism of fatty acid,108 and energy production.
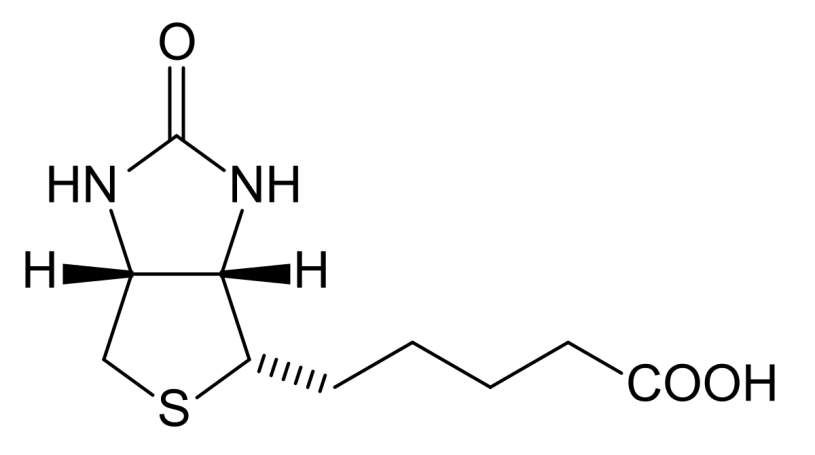
Figure4‑8. Structure of biotin
The biotin receptors expression levels are even higher than folate and B12 receptors in the following cancer cells: L1210FR (leukemia), ID-8 (ovarian), Colo-26 (colon), P815 (mastocytoma), M109 (lung), MMT06056 (breast), and so on. 106 Thus, biotin receptor is a great target for tumor targeting drug delivery.109 Previously, Ojima group have utilized it in several drug conjugates and validated the drug release by fluorescent molecular probes.104 Also, our group member Dr. Vineberg identified MX-1 and MCF7 as BR++ and BR+++ cell lines respectively, in addition to the cell lines Russell-Jones reported.
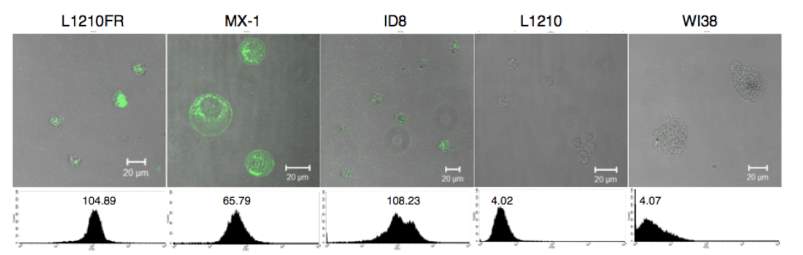
Figure 4‑9. CFM image and flow cytometry analysis of biotin receptor overexpressing cell lines
Ojima group monitored internalization of biotin receptor-mediated endocytosis (RME) and validated biotin tumor targeting drug delivery system by confocal fluorescence microscopy (CFM) and flow cytometry.104 Comparing the fluorescence signal after incubating biotin–fluorescein with L1210FR cells (BR+++) at 37 oC and 4 oC for 3 hours found decreasing signal in cells incubated at 4 oC, which indicated inhibited cellular uptake at lower temperature104 and proved the internalization of biotin-fluorescein is an energy depending endocytosis procedure. Preincubated L1210FR cells with excess biotin molecule for 1 hour and followed by addition of biotin–fluorescein resulted in total absence of fluorescence in CFM image.104 This phenomenon confirmed the internalization of biotin-fluorescein is mediated by biotin receptor endocytosis. Another probe: biotin–linker– taxoid–fluorescein was applied to validate the release of drug. After incubating the biotin-linker-taxoid-fluorescein probe with L1210FR cells (BR+++) at 37 oC for 3 hours, it underwent same internalization manner with biotin-fluorescein probe.104 Glutathione ethyl ester was added and coincubated with cells for another 1 hours. The CFM image revealed fluorescence-labeled microtubule bundles suggesting free fluorescent taxoid binds to the microtubules.104 The existence of free fluorescent taxoid confirmed the self-immolative linker was cleaved by glutathione to release drug and thiolactonization took place as expected.
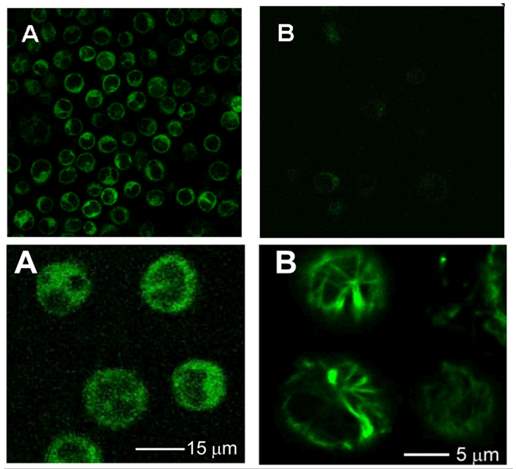
Figure 4‑10. CFM image of biotin-fluorescein probe in L1210FR cells.
Upper A, biotin-fluorescein incubate at 37 oC; Upper B, biotin-fluorescein incubate at 4 oC; Lower A, biotin-linker-taxoid-fluorescein incubate at 37 oC; Lower B, biotin-linker-taxoid-fluorescein with glutathione ethyl ether incubate at 37 oC
These experiments concluded that biotin targeting strategy works well for cancer cells overexpressing biotin receptor, the biotin drug conjugate would internalize through biotin receptor mediated endocytosis. The release of the therapeutic drug is triggered by glutathione via cleavage of disulfide linker and thiolactonization.
4.1.5 Self-immolative disulfide linker
In the tumor targeting drug delivery system, the drugs’ activity and toxicity are masked by “prodrug deactivation”.110 After internalization into cancer cells or tumor microenvironment, linker will be cleaved and release cytotoxic drug. Thus, the linker should be stable in normal tissue or circulation system to prevent unnecessary toxicity and irreversibly cleaved in tumor to release drug. And the linker should be bifunctional to connect with warhead and targeting molecule by two ends. Conjugation of drug and tumor targeting moieties by ester bond is unstable and can be easily hydrolyzed, while conjugation by amide or ether bond is too stable. Meanwhile, introducing crosslinkers need orthogonal functional groups which do not exist in drug.110 Hence, a trigger unit, such as cleavable peptides, reduction-cleavable disulfides, or acid-sensitive hydrazine groups is introduced into the drug conjugates.110
Tumor microenvironment is acidic with pH of 6.5-6.9, and it overexpresses certain proteases like MMPs.111 Endosomes and lysosomes, where the drug conjugate will locate after internalization in the cells, has an acidic environment with pH of 4.5-5.5,112 and a high concentration of reducing agents such as glutathione, and proteases like cathepsin. These properties could be exploited to design “triggers” only respond to these environment.
Self-immolative disulfide bonds were designed based on the high intracellular concentration of glutathione. Disulfide bond could be reduced by glutathione in the cells. Glutathione’s concentration is found to be 1000 times greater in tumors (2-8 mM) than in blood plasma (1-2 µM).113 The dramatic difference of glutathione concentration between tumor tissue and normal tissue enables disulfide linkers’ stability in normal physiological environment and rapidly cleaveage inside cancer cells.
However, simple disulfide bond breakage did not necessarily release free drug, but usually compromisingly modified drug with linker pieces. Energetically favorable cyclization strategies such as formation of five or six membered lactones, lactams would be applied to release free drug.114 Here, a phenyl group was introduced next to the disulfide bond. After reduction by glutathione, the free thiols would undergo nucleophilic attack to the ester bond and release drug through thiolactonization.114 Also, a methyl substituent was incorporated proximal to the disulfide bond to lengthen the linker’s circulation time.
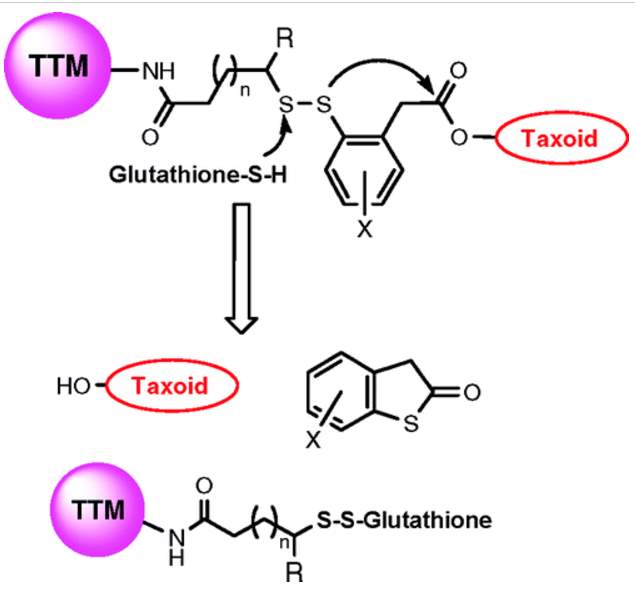
Figure 4‑11. Mechanism of drug release of disulfide linker
The drug release mechanism of disulfide linker has been proved by time-resolved 19F NMR in a model system of fluorine-labeled taxoid.115 The release of drug followed a two-step manner. The reference signal of F in C2-benzoate was stable at −112.5 ppm with no chemical shift change with time. After glutathione addition for 1 hour, the drug-linker conjugate was reduced to 2’-fluoro(sulfhydryl)phenylacetyltaxoid (3-A) first, with a F signal at −119.2 ppm, instead of direct formation of the thiolactonization product 3-B. Increasing formation of 3-B was observed with decreasing amount of 3-A, over the time course. 3-A was a key intermediate involves in the 2-step self-immolation process.114
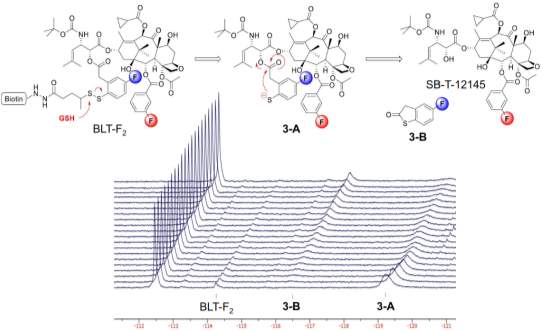
Figure 4‑12. 19F NMR for the disulfide linker cleavage and thiolactonization process of BLT-F2
4.2 Design and Synthesis of SB-T-1214 and Biotin functionalized gold nanoparticles
4.2.1 Design of SB-T-1214 and Biotin functionalized gold nanoparticles
As mentioned above, nanoparticles internalization by passive targeting has some limitations in targeting tumor tissue efficiently. Cancer cells could reduce their interaction with nanoparticles and inhibit the internalization of nanoparticles; especially they can downregulate the expression of opsonins to prevent the internalization of PEGylated nanoparticles.116 Lack of further control might result in undesired drug release which reduces therapeutic efficiency. 117 A feasible strategy to overcome these limitations is to attach targeting moieties onto the surface of nanoparticles. In this way, the nanoparticles will be accumulated in the tumor tissue by the enhanced permeability and retention (EPR) effect in a passive targeting way. And the targeting moieties on the surface of these nanoparticles would benefit their internalization into the cancer cells through receptor mediated endocytosis (RME) in an active targeting way. By combining passive targeting (EPR effect) and active targeting (RME) together, the drug delivery system will achieve high tumor-specificity and drug-delivery efficiency.118 Some of these drug delivery systems are under clinical trials, including transferrin receptor targeted liposome for delivery of oxaliplatin (MBP-426, phase 2)119, prostate-specific membrane antigen PSMA targeted (via ACUPA) polymeric nanoparticle for delivery of docetaxel (BIND-014, phase 2)120, and so on. Building active targeting nanoparticles is a promising approach to specifically deliver therapeutic drugs to cancer cells. However, although several antibodies or proteins have been applied as targeting moieties and reached clinical trials, small tumor targeting molecules have not succeeded in this approach.121
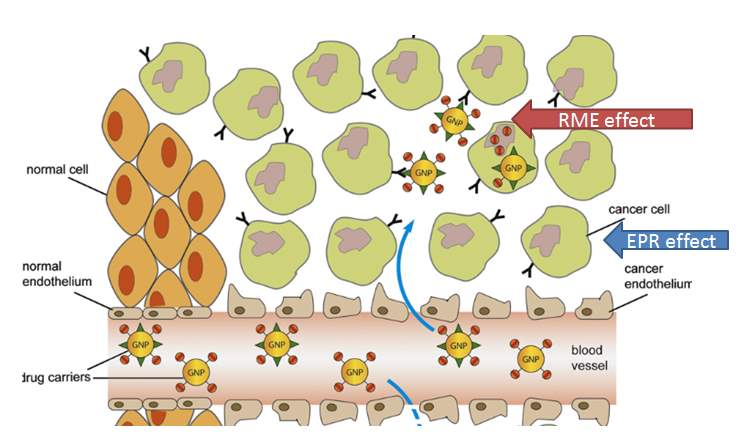
Figure 4‑13. EPR and RME effect of targeted nanoparticle drug delivery system
Gold nanoparticles (Au NPs) have superior advantages in the size, stability and biocompatibility, and it is a potential drug delivery system. There are several advantages in using Au NPs. The gold core is inert and low toxicity,73 and it has a wide range of diameter from 1 to 150 nm.
Here, I designed the biotin and SB-T-1214 functionalized gold nanoparticles as drug delivery system. Two approaches for targeting –“passive targeting” ERP effect and “active targeting” RME effect would be utilized at the same time. The function of Au NPs is to anchor the delivery system’s arrival at the tumor tissues through leaky blood vessel after administration into circulatory system. Small tumor targeting molecules have their advantages in stability, ease of production, low cost, and assumable high yield of conjugation. Biotin (vitamin H) is a water soluble growth promoter for cells. Due to rapid proliferations of cancer cells, they often over-express biotin-specific receptors on the cell surface. Biotin will serve as a targeting molecule to bind with the biotin receptor expressed on the surface of cancer cells, and lead to endocytosis of delivery system in an “active targeting” way. SB-T-1214, a second generation of taxoids, exhibits great potency and anti-MDR activity towards cancer cells, has been chosen as warhead.
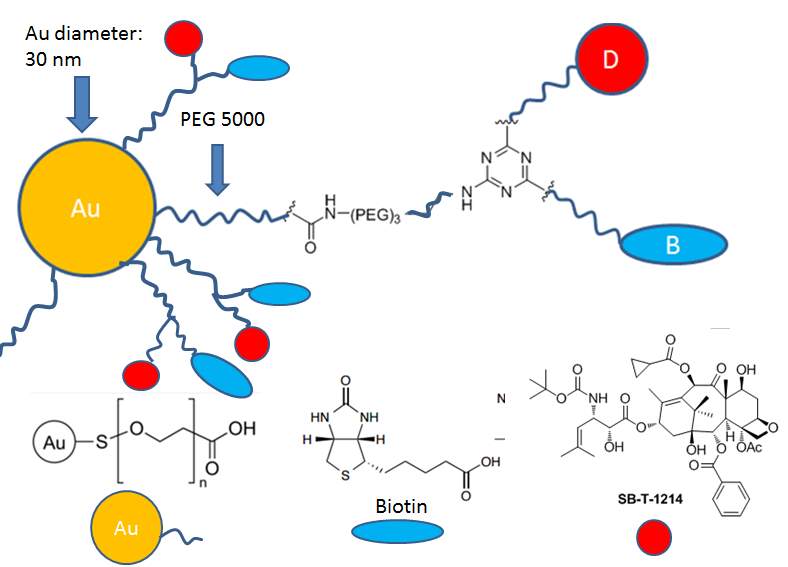
Figure 4‑14. Scheme of biotin and SB-T-1214 functionalized gold nanoparticles
PEGylated AuNPs and PEG chain length
Gold nanoparticles in drug delivery system are usually grafted with a surface stabilizer to prevent Van der Waals (VDW) and depletion forces resulted aggregation of the gold.122, 123 The surface of gold is usually grafted with PEG polymers to improve water solubility, biocompatibility and avoid oponization in circulation system.124 Polyethylene glycol (PEG) coating can reduce the surface charge (zeta potential) of Au NPs to -40 ~ -10 mV, and thus improve blood stability.125 The PEG chain is introduced by stable gold-thiol bond.126 The PEG coated Au NPs could be monodisperse distributed in the physiological environment, and they show long-term blood circulation and long blood half-life.123 Moerover, PEGylated gold nanoparticles could be functionalized with different groups including amine, carboxylic acid, biotin, etc. on the other end to facilitate its further conjugation with drug.
The PEGylating density (σp) and PEG polymerization degree (N) significantly affect cellular uptake of the PEGylated gold nanoparticles.127 When PEG density σp is small, the cellular uptake efficiency is diminished. The PEG polymerization degree N plays an important role in internalization rate and surface free energy barrier.127
After the gold nanoparticles enter the biological milieu, they are likely to absorb the serum proteins to form a protein corona surrounding them.128 Generally, gold nanoparticles enter cells through endocytosis pathway. Three different proteins (clathrin-, caveolin-, or scavenger- receptors) mediated endocytosis involve in the procedure depending on the gold nanoparticles’ size and cell type.129 For 20 nm PEGylated gold nanoparticles, caveolin receptor mediated endocytosis is the major internalization pathway, while for 50 nm ones, clathrin receptor dominate the internalization.129 Presence of protein corona reduces gold nanoparticles’ interaction with cell membrane and thus prevents AuNPs’ adhesion and cellular uptake. Increasing the PEG grafting density could inhibit serum protein adsorption.130
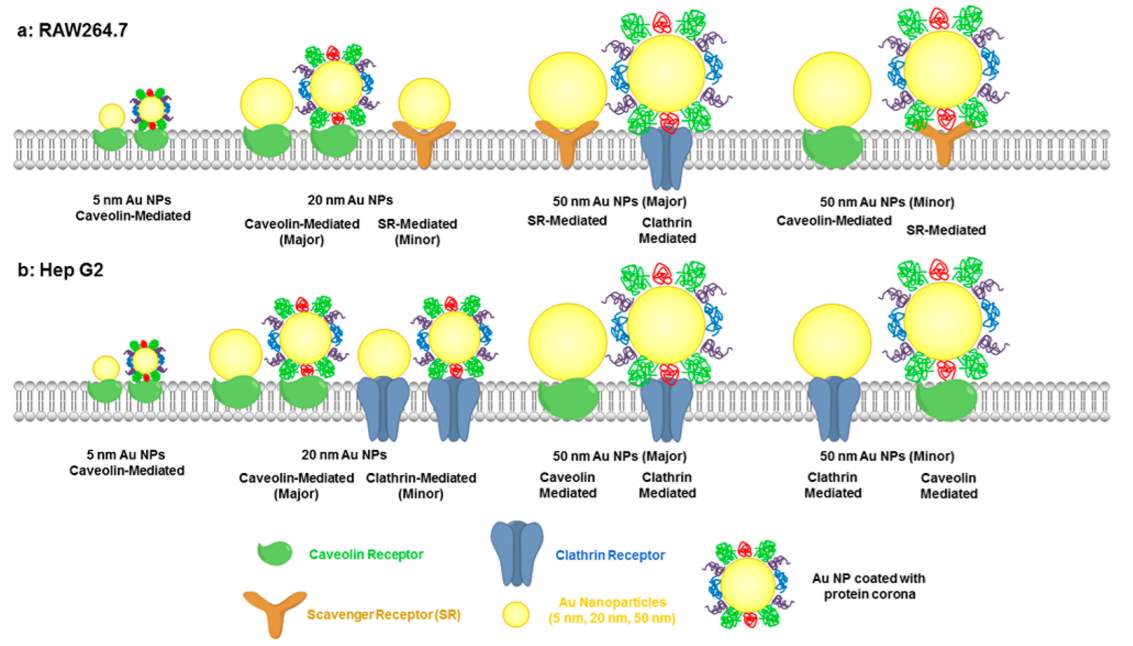 Figure 4‑15. Internalization of PEGylated gold nanoparticles
Figure 4‑15. Internalization of PEGylated gold nanoparticles
Shape of PEGylated AuNPs changed with coating PEG chain length. Gold nanoparticles coated by shorter PEG chain (flory radius smaller than the distance between two inserted PEG) would apply brush like conformation, in which all the terminal is extended toward outside. If gold nanoparticles are coated with longer PEG chain (flory radius is larger than distance between two inserted PEG), they will show mushroom conformation.76 Significant differences between nanoparticles with the two PEG conformations in terms of binding affinity with serum proteins have been observed. Brush NPs showed less binding of serum proteins than mushroom NPs on average.131 According to the Flory radius calculated by the formula (F=αn3/5), PEG5000 has a radius of 6.27 nm while PEG2000 has a radius of 3.46 nm.
Gold nanoparticle size and charge
To capitalize on the EPR effect, and to evade RES capture, a drug carrier must be in a narrow size range from approximately 10 nm to 100 nm.132,133 Generally, drug carriers smaller than 10 nm are likely to be filtered or cleared by kidney.134 Nanoparticles in the size of 20–200 nm are able to extravagate through leaky tumor vascular fenestrations by EPR effect and escape filtration by liver and spleen. However, larger carriers with size of 100~200 nm are easily to be cleared by reticulo-endothelial system (RES)135 and lead to toxicity.126 Especially for NPs with size up to 150 nm, they are entrapped in liver and spleen.133 Thus, the preferential size of nanoparticles to achieve EPR effect is narrowed to 10 ~100 nm.
The size of nanoparticles effects their distribution in the body and cellular uptake. Large size NPs (>100 nm) tend to accumulate in spleen and liver while small size NPs (< 10 nm) are likely to be filtered by kidney. Spleen, liver and kidney were the main metabolism organs for gold nanoparticles. In Zhang’s research of biodistribution of PEGylated gold nanoparticles,136 the 5 nm ones had a wide distribution in live, heart, kidney, while the 10 and 30 nm particles preferentially stayed in the liver and spleen, respectively. To assess the toxicity, some biochemistry and hematology assays have been conducted. The results clearly showed that the 10 nm particles are highly toxic to the liver and that the 60 nm particles are toxic to both the kidney and liver. However, the 5 nm and 30 nm particles did not cause any significant liver and kidney damage.136 Thus, I decide to use the gold nanoparticles with 30 nm diameters for the construction of my drug delivery system.
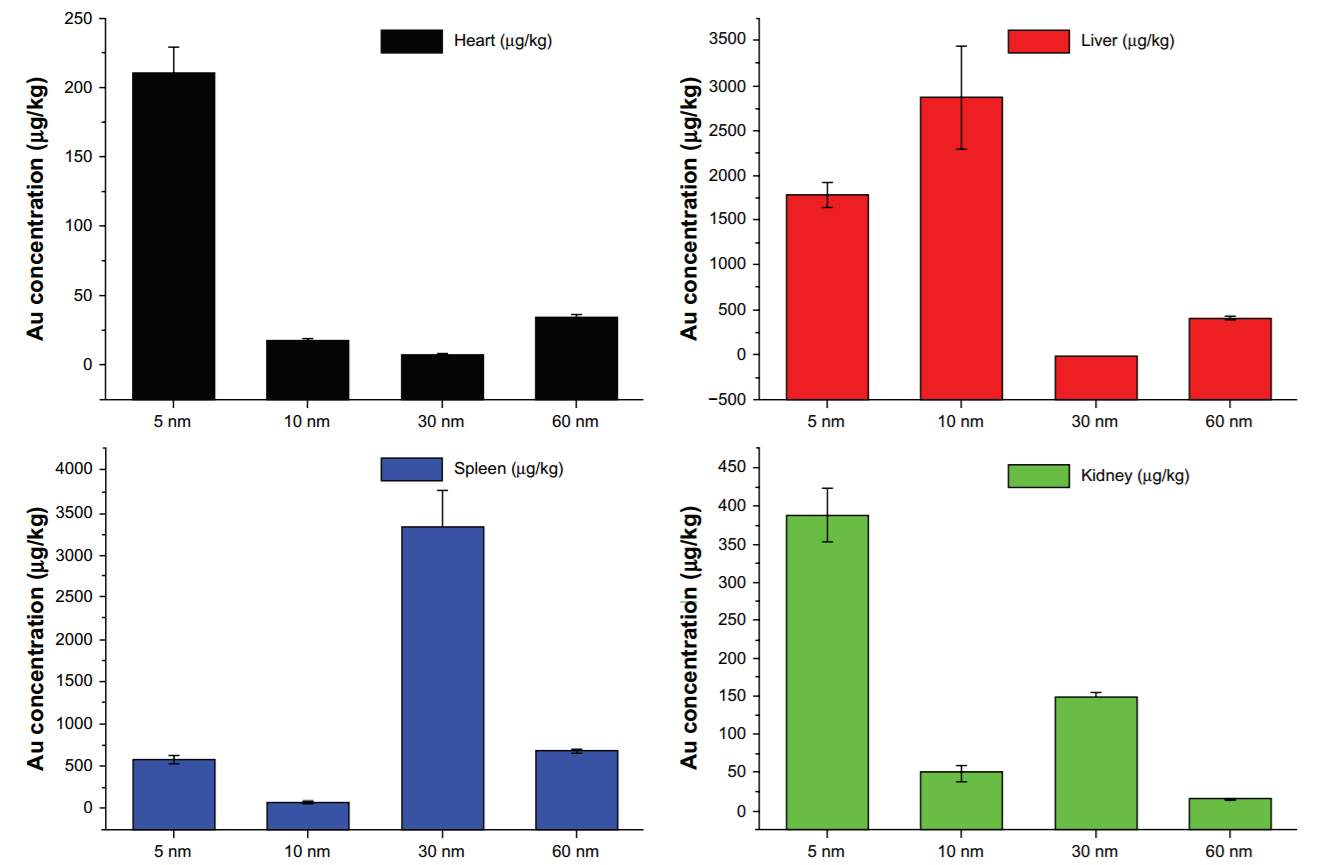
Figure 4‑16. Biodistibution of Au NPs with different sizes
Also, the gold nanomedicine CYT-6091 under phase 1 clinical trial investigation also uses the gold core with diameter of 28 nm, ~30 nm. Similarly, they coated the gold by PEG chain, therapeutic agent, and tumor necrosis factor-α (TNFα) targeting ligand. With PEG coating and TNFα targeting molecule, CYT-6091 showed high tumor specificity in the in vivo study as shown below. Upon administration, the gold nanomedicine accumulated in liver and lung, and after 3 hours, half of them were cleared from healthy tissue, and stayed in tumor.137
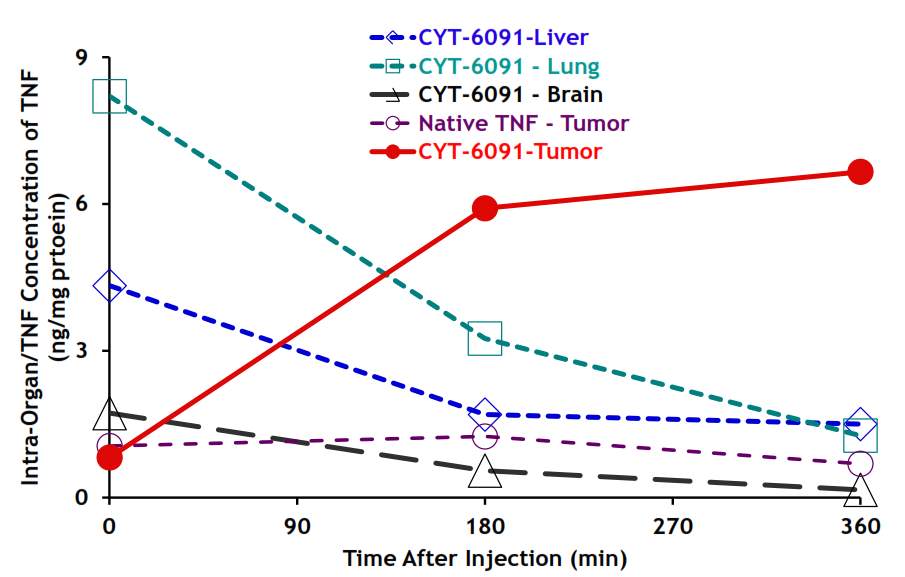
Figure 4‑17. Biodistribution of CYT-6091 in vivo
Charge of the gold nanoparticles is essential for their cellular uptake, and thus will influence opsonization, circulation time and interaction with macrophages. Positively charged nanoparticles are more likely to be uptake by macrophages in lung, liver and spleen than negatively charged ones.138 Coating the gold nanoparticles by PEG chain converted the charge from positive to negative (-40 ~ -10 mV) and thus solved the problem.
To sum up, gold nanoparticles (30 nm) with PEG5000 or PEG2000 coating was selected for construction of drug delivery system. The terminal functional group of PEG was carboxylic acid to facilitate further drug and biotin loading, and eventually, the unreacted carboxylic acid will be capped by hydrazine benefit the membrane transportation. The biotin was introduced as tumor targeting molecule and SB-T-1214 was selected as warhead. 1,3,5-triazine served as a three-arm splitter, one arm was connected with a polyethylene glycol spacer and a biotin to improve the solubility and facilitate biotin’s exposure; another arm was conjugated with self-immolative disulfide linker and warhead SB-T-1214 to achieve facilely and rapidly drug release; the third propargylamino arm was used for attachment of a polyethylene glycol spacer and gold nanoparticle through EDC coupling reaction to improve solubility and extend the biotin-drug core into the outer space. This biotin and SB-T-1214 functionalized gold nanoparticle drug delivery system is expected to demonstrate efficient internalization, high-specific targeting, extended accumulation, and great cytotoxicity.
Figure 4‑18. Scheme presentation of Au NPs drug delivery system
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


Related Content
All TagsContent relating to: "Medical Technology"
Medical Technology is used to enhance the medical care and treatment that patients are given in healthcare settings. Medical Technology can be used to identify, diagnose and treat medical conditions and illnesses.
Related Articles
DMCA / Removal Request
If you are the original writer of this dissertation and no longer wish to have your work published on the UKDiss.com website then please: